
Amyotrophie spinale
ou atrophie musculaire spinale
L’amyotrophie spinale (SMA, Spinal Muscular Atrophy) est une des maladies génétiques rares les plus fréquentes. Transmise sur le mode autosomique récessif, elle affecte approximativement 1 sur 6000 à 1 sur 10000 naissances et est responsable d’une partie significative de la mortalité infantile. Cette maladie résulte de la dégénérescence des motoneurones alpha issus de la corne antérieure de la moelle épinière (Dowling 2017). Comme on peut le voir sur le schéma ci-dessous (Figure 1), ces neurones assurent l'innervation des muscles squelettiques du tronc et des membres. La perte de ces motoneurones a pour conséquences évidentes une atrophie et une faiblesse musculaire plus ou moins sévères (Kobl 2015; Prior 2010; Verhaart 2017). Le développement moteur est évalué par un score sur l’échelle CHOP INTEND (Children Hospital of Philadelphia Infant Test of Neuromuscular Disorders). Une étude réalisée sur 34 nourrissons SMA-I a montré qu’un seul d’entre eux atteint un score de 40 points après 6 mois (Finkel 2014). Généralement le score de CHOP INTEND décroît de 10,7 points en moyenne entre 6 et 12 mois.
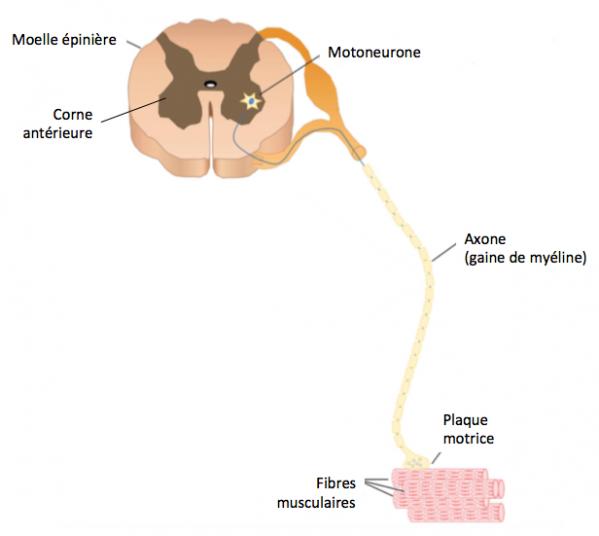
Figure 1. Schéma d'un motoneurone (adapté de Dowling 2017)
Les divers types de la maladie
En 1991, devant l’hétérogénéité clinique de cette maladie, une classification a été mise en place et basée sur les niveaux de fonction motrice (enfants pouvant supporter une position assise, ou la marche, test CHOP INTEND) et l’âge d’apparition des symptômes (Kobl 2015). Il existe 4 types de SMA si on exclue le stade fétal.
SMA-0 (stade fétal) : ce type de SMA est utilisé pour décrire les nouveaux nés qui présentent de sévères hypotonies avec une histoire d’absence de mouvements durant la vie fétale. Dans ces cas, la maladie est probablement mise en place en période prénatale. La détresse respiratoire et l'insuffisance cardiaque représentent les problèmes majeurs chez ces enfants dont la plupart ne survie pas au delà de 6 mois.
SMA-I (infantile, aussi appelée maladie de Werdnig-Hoffman) : apparaît avant 6 mois avec hypotonie musculaire sévère notamment des muscles intercostaux, absence de contrôle de la tête, réflexes tendiniques absents ou faibles, faiblesse de la langue et difficultés de déglutition et de succion, détresse respiratoire avant 2 ans. Ces enfants ne peuvent pas se tenir assis ni marcher. Pas d’altération des capacités cognitives. Age moyen de survie et/ou d’assistance respiratoire: 10 mois et demi. Soixante pour cent des malades atteints de SMA sont de type I.
SMA-II (intermédiaire, aussi appelée maladie de Dubowitz): Ces enfants pourront se tenir assis à un certain moment de leur développement, mais ne pourront jamais marcher sans assistance. Cette forme intermédiaire de la maladie se manifeste de façon progressive au niveau des membres inférieurs, la faiblesse des membres supérieurs étant moins marquée. La plupart des problèmes résultent de complications orthopédiques (os et cartilages) avec scoliose progressive, contractures articulaires et ankylose. Une faiblesse des muscles intercostaux associée à la scoliose peut conduire à des problèmes respiratoires. Pas d’altération des capacités cognitives.
SMA-III (juvénile, aussi appelée maladie de Kugelberg-Welander) : Les enfants et adultes pourront marcher sans assistance à une certaine période de leur vie. Mais ils présentent une faiblesse progressive des membres inférieurs (les membres supérieurs étant moins affectés) pouvant conduire à l’obligation d’utiliser un fauteuil roulant. Ces patients ne présentent pas de scoliose et pas ou peu de faiblesse des muscles respiratoires. Pas d’altération des capacités cognitives et durée de vie normale.
SMA-IV (adulte): les patients atteints par ce type de SMA, la forme la moins sévère de la maladie, représentent moins de 5% des cas. Ils sont libres de tout mouvement et la maladie ne se manifeste que durant l’âge adulte, le plus souvent après 30 ans.
Les gènes SMN1 et SMN2
Le gène responsable de la maladie est SMN1 (survival of motor neuron 1, ou survie du motoneurone 1) porté par le chromosome 5 et dont la perte de fonction (délétion totale du gène ou mutations rendant la protéine inactive) conduit à la mort des motoneurones de la moelle épinière, avec pour conséquence l’atrophie musculaire, la destruction de jonctions neuromusculaires et la paralysie (voir Figure 1).
Le gène SMN1 est très conservé et présent dans le génome de tous les eucaryotes. Chez l’homme, cependant, une duplication génomique d’une longueur de 500kb environ dans la région q13.2 du chromosome 5 a donné naissance à un second gène survival of motor neuron, SMN2. Les deux gènes se trouvent de ce fait dans deux régions différentes du chromosome 5, SMN1 dans la région télomérique et SMN2 dans la région centromérique. Ils sont 99% homologues, s’étendent sur 27kb et possèdent 9 exons (1, 2a, 2b, 3-8). L’ARNm de 1,7kb code pour une protéine de 294 acides aminés (Lefebvre 1997; Wirth 2000; Prior 2010).
La protéine SMN (aussi appelée SMA) présente dans le cytoplasme et le noyau des cellules joue un rôle apparemment essentiel dans le métabolisme des ARNmessagers, en particulier sur l’épissage par interaction avec la protéine snRNP (petite ribonucléoprotéine nucléaire). La protéine SMN est présente dans les axones des motoneurones et d’autres fonctions anti-apoptotique et activation transcriptionnelle lui sont suspectées (Harada 2002; Li 2017).
Bien que très proche, les deux gènes SMN1 et SMN2 présentent des différences dans leur séquence nucléotidique, une dans l’intron 6, une dans l’exon 7 (substitution C-->T, silencieuse), deux dans l’intron 7 et une dans l’exon 8 (région non traduite). Les deux protéines codées par SMN1 et SMN2 sont donc identiques. Cependant, la substitution C-->T dans l’exon 7 de SMN2 revêt une importance cruciale. Présente chez tous les individus, elle conduit à un épissage aberrant de l’exon 7 et à la production d’une protéine SMN incomplète (SMN∆7) et instable, et donc inactive (Figure 2) (Kobl 2015; Lefebvre 1997; Wirth 2000; Finkel 2014).
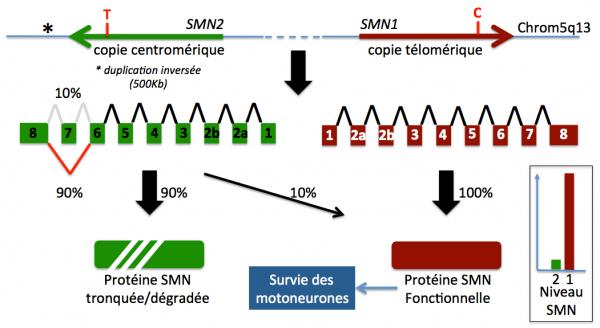
Figure 2. Schéma du locus SMN sur le chromosome 5 région q13 chez un sujet non porteur (adapté de Kolb 2015). En haut, les deux gènes SMN1 et SMN2. Les nucléotides C et T (mutation T->C) sont représentés en rouge. La partie centrale montre l'impact de cette mutation sur l'épisage de l'ARN SMN2 avec perte de l'exon 7. La partie basse de la figure montre la conséquence de cette mutation sur la production de la protéine. SMN1 normal code pour la protéine SMN fonctionnelle, alors que SMN2 ne code que pour 10% environ de la protéine normale, la majeure partie de la protéine codée par ce gène étant tronquée (délétion de l'exon 7) et donc instable et inactive. Au final, le niveau de protéine SMN est donc la somme de la protéine issue du gène SMN1 et SMN2.
En bref donc, le gène télomérique SMN1 code pour la protéine complète tandis que le gène SMN2 produit la protéine incomplète (SMN∆7, délétion de l’exon 7) inactive. Toutefois, le gène SMN2 génère une petite quantité (environ 10%) de la protéine SMN normale.
SMN2 n’est pas responsable de la maladie, mais nous verrons que sa contribution, bien que limitée (environ 10%) à la production de la protéine SMN normale, joue un rôle majeur sur le phénotype des malades atteints de SMA (Figure 3). En effet, dans les cas où le gène SMN1 est absent (délétion homozygote) ou muté (mutations inactivatrices de la protéine), la seule contribution à la production de la protéine SMN complète est assurée par le gène SMN2 (environ 10% de sa production totale).
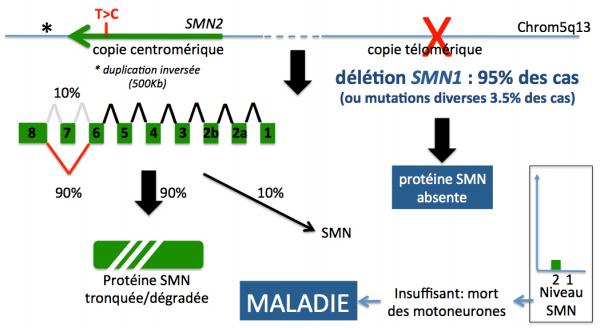
Figure 3. Schéma du locus SMN sur le chromosome 5 région q13 chez un sujet malade (adapté de Kolb 2015). Dans ce cas, seule la contribution du gène SMN2 est responsable du niveau de la protéine SMN, le gène SMN1 étant soit absent, soit muté.
Génotypes de la SMA
Plusieurs copies des gènes SMN1 et SMN2 peuvent être présentes dans le génome humain (Figure 4) (Verhaart 2017). Généralement les individus normaux (non porteurs) présentent 2 ou 3 copies de SMN1 non muté dans leur génome (par exemple une copie sur chaque chromosome 5 ou 2 copies sur un chromosome 5 et une copie sur l’autre). Les porteurs (non malades) peuvent avoir au moins une copie (ou deux sur le même chromosome) du gène SMN1 non muté avec une ou deux copies mutées sur l’autre chromosome. On estime que chez les caucasiens environ 1 personne sur 50 est porteuse.
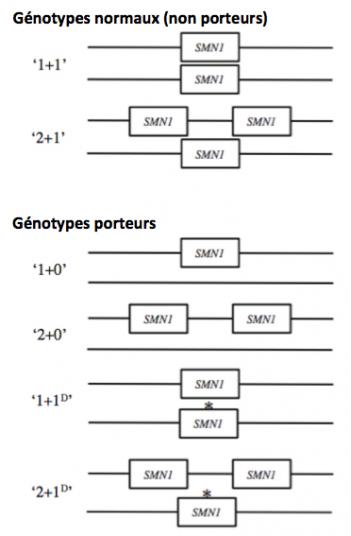
Figure 4. Génotype des sujets non porteurs (normaux) et porteurs (Adapté de Verhaart 2017). A chaque génotype est associé un terme d'identification prenant en compte la présence ou l'absence et le nombre de copie du gène SMN1. Par exemple: "1+1" signifie que les deux chromosomes 5 sont chacun porteur d'une copie de SMN1; autre exemple: "2+1D" signifie qu'un des chromosomes 5 du sujet porte 2 copies de SMN1 alors que ce gène est muté sur l'autre chromosome 5 (D: microdélétion). Les astérisques (*) figurent les cas où le gène SMN1 est présent mais muté et donc non codant pour la protéine SMN. Le nombre 0 signifie que le gène SMN1 est absent sur le chromosome (exemple: "2+0" soit deux copies de SMN1 sur un chromosome 5 et 0 sur l'autre).
Le gène SMN1 est absent ou muté chez tous les malades atteints de SMA (aucune copie du gène normal)
Dans ces cas, la sévérité de la maladie corrèle avec le niveau de protéine SMN codée par SMN2. Ainsi SMN2 agit comme un modulateur de la sévérité de la maladie car le nombre de copies de ce gène varie selon les individus. Plus le nombre de copies de SMN2 est élevé, plus la quantité de protéine SMN fonctionnelle est élevée et moins sévère est la maladie (Table 1) (Bowerman 2017).
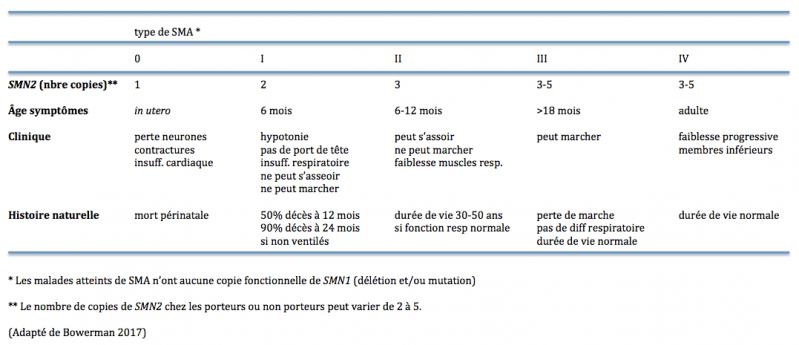
Table 1. Caractéristiques cliniques et moléculaires des divers types de SMA.
Traitements
Il n’y a pas de traitement pour cette maladie excepté les traitements symptomatiques (assistance respiratoire ou appareillage, par exemple). Récemment deux approches thérapeutiques ont été testées avec un certain succès, même si il reste encore beaucoup de progrès à réaliser.
Oligonucléotide anti-sens : le Nusinersen
Une des stratégies thérapeutiques est focalisée sur l’augmentation de l’expression de la protéine SMN à partir du gène SMN2 (le seul présent chez les malades les plus sévèrement touchés et chez qui SMN1 est absent ou muté). Dans ce but un oligonucléotide anti sens (le nusinersen, Spinraza®, Ionis Pharmaceuticals/Biogen) a été développé afin d’inhiber l’épissage aberrant de l’exon 7 dans l’ARN pré-messager de SMN2. Cet oligonucléotide a montré des effets bénéfiques au niveau musculaire dans un modèle de souris atteinte de SMA sévère, avec une augmentation de durée de vie des animaux de 16 à 25 jours (DiMatteo 2008; Hua 2010).
En Décembre 2016 le nusinersen a été approuvé par la FDA aux États Unis pour le traitement de la SMA-I. Une étude récemment publiée sur des enfants atteints de SMA-I et traités avec le nusinersen a donné des résultats encourageants (Finkel 2017 ; publié le 2 Novembre 2017; Finkel 2016). Voici un résumé de cette étude.
Protocole clinique
Dans cette étude, 122 nourrissons dont les symptômes avaient été détectés dès, ou avant, 6 mois ont été enrôlés. Ces enfants ont été génotypés et portaient deux copies du gène SMN2 mais aucune de SMN1. Ils ont été répartis en deux groupes, un groupe recevant le nusinersen par administration intrathécale par voie lombaire (aux jours 1, 15, 29, et 64, et des doses de maintenance aux jours 183 et 302), et un groupe contrôle pour lequel une simulation d’injection était pratiquée afin de ne pas influencer les familles ni les personnels médicaux (médecins et infirmiers) chargés de l’évaluation du traitement. Précisons ici que ces personnels ne savaient pas à quel groupe (nusinersen ou contôle) appartenaient les enfants qu’ils évaluaient. Seuls les personnels traitants connaissaient cette répartition.
Résultats
Dans le groupe nusinersen les enfants ont montré des améliorations de leur activité motrice très largement supèrieure à ceux du groupe contrôle (41% contre 0%). A la fin de l’analyse, ce pourcentage était de 51%. Cependant, seulement 8% de ces enfants pouvaient s’asseoir de façon autonome et seulement 1% pouvaient se lever. A la fin de l’étude, 39% des enfants du groupe nusinersen et 68% du groupe contrôle étaient soit décédés soit placés sous assistance respiratoire permanente. Les meilleurs résultats ont été observés sur les enfants ayant été traités au maximum 13 mois après détection des symptômes de la maladie.
Depuis la fin août 2017, le nusinersen (Spinraza) peut temporairement être prescrit et remboursé en France et en Europe (EMA, European Medical Agency) pour toutes les personnes atteintes de SMA-I et -II. Ce type de traitement reste toutefois très invasif.
Thérapie génique
Tout récemment, une thérapie génique de la SMA-I a donné des résultats très encourageants (Mendell 2017 ; publié le 2 Novembre 2017). Voici un résumé de cette étude.
Protocole clinique
Dans cette étude, 15 nourrissons SMA-I ont reçu une thérapie génique. Ces enfants ont été génotypés et portaient deux copies du gène SMN2 mais aucune de SMN1. Cette thérapie a consisté en une injection intraveineuse d’un virus adénoassocié de sérotype 9 (AAV9, non toxique) portant un ADN complémentaire SMN1 codant pour la protéine SMN manquante. Ce virus est nommé AVXS-101 et développé par la compagnie AveXis. Dans une première cohorte (cohorte 1) trois enfants (âge moyen 6,3 mois) enrôlés entre Mai 2014 et Septembre 2014 ont reçu une faible dose du virus (6,7x1013 virions par kilo). Dans une deuxième cohorte (cohorte 2) 12 enfants (âge moyen 3,4 mois) enrôlés entre Décembre 2014 et Décembre 2015 ont reçu une plus forte dose du virus (2x1014 virions par kilo).
Résultats
Au 7 Août 2017, date de fin de l’étude, les 15 enfants SMA-I étaient en vie et avaient atteint l’âge de 20 mois (au moins), alors que normalement seulement 8% d’entre eux (c’est à dire un seul enfant sur les 15) auraient dû être encore en vie. Un seul d'entre eux nécessitait une ventilation permanente, alors que cette assistance est normalement requise au delà de 10 mois et demi, chez tous les enfants non traités.
Le score CHOP INTEND de tous les enfants des cohortes 1 et 2 a augmenté et/ou s’est maintenu (pas de décroissance) durant l’étude. Aucun des enfants de la cohorte 1 n'a pu atteindre une activité motrice suffisante et un d'eux a dû avoir recours à une assistance respiratoire. Dans la cohorte 2, 11 enfants ont atteint un score supérieur à 40 points (Figure 5). L’augmentation était moindre chez les enfants de la cohorte 1, vraisemblablement du fait de la plus faible dose d’adénovirus administrée. Toujours dans la cohorte 2, les enfants ont montré de réels progrès dans les fonctions motrices avec 9 enfants capables de garder une position assise pendant au moins 30 secondes, 9 contrôlant le port de tête et 2 capables de ramper, de se lever et marcher sans assistance. Onze de ces enfants ont pu parler. A noter que chez les enfants SMA-I non traités, aucune des “performances” évoquées ci-dessus n’est atteinte. Dix des 12 enfants de la cohorte 2 n’ont pas eu besoin d’assistance respiratoire, 11 d’entre eux ont acquis la capacité d’avaler et 4 ont été capables de se nourrir seuls.
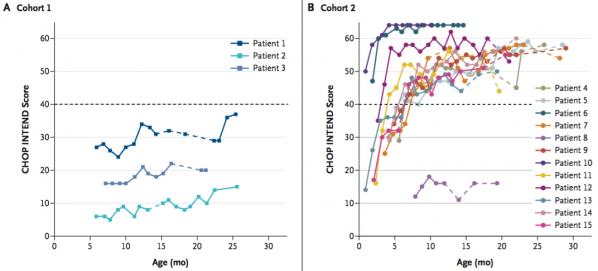
Figure 5. Evolution du score de CHOP INTEND (activité motrice) chez des nourrissons ayant été traités par thérapie génique (Source: Mendell 2017). On voit que les enfants de la cohorte 2 (forte dose de virus) progressent mieux que ceux de la cohorte 1 (faible dose de virus). La plupart des enfants de la cohorte 2 ont atteint un score supérieur à 40 après 10 mois.
Les effets secondaires de ce traitement par thérapie génique avec AAV9 ont été limités à une augmentation des transaminases chez 2 enfants après 3 semaines de traitement, sans autre anomalie enzymatique. Cette augmentation des transaminases a été atténuée par un traitement à la prednisolone (corticoïde) qui a ensuite été étendue aux autres enfants.
En conclusion
Chez des nourrissons atteints de SMA-I, une administration unique d’un virus adénoassocié de type 9 (haute dose) portant un ADN complémentaire SMN1 codant pour la protéine SMN manquante a permis un allongement de la survie, une amélioration des fonctions motrices et une augmentation du score CHOP INTEND à un niveau jamais atteint pour cette maladie. A la date de cette publication et après deux ans de suivi, aucune diminution/régression dans les paramètres cliniques et les fonctions motrices n’a été observée.
(Étude réalisée par les chercheurs du Center for Gene Therapy at the Research Institute at Nationwide Children’s Hospital et le Departments of Pediatrics, Neurology, Pathology et Molecular and Cellular Biochemistry Ohio State University. Co-financement par AveXis, Bannockburn, IL; ClinicalTrials.gov number, NCT02122952).
Discussion de ces résultats
Il n’est pas aisé de comparer les résultats de ces deux études du fait d’un protocole totalement différent et du fait aussi de l’âge différents des enfants traités (3,4 mois avec l’AAV9 contre 5,4 mois avec le nusinersen).
Un avantage évident de la thérapie génique par AAV9 est le fait qu’elle ne requiert qu’une administration du virus par voie intraveineuse, alors que la thérapie via le nusinersen se fait par des injections répétées (délicates et invasives) par voie intrathécale lombaire.
Dans les deux études, le suivi a été limité et la durabilité de l’effet reste incertaine. Si l’expression de SMN1 par thérapie génique avec AAV9 décline avec le temps, il ne sera pas possible de répéter le traitement car des anticorps anti-AAV9 (protéines de capside du virus) ont été forcément produits. De plus, l’impact de ces deux thérapies sur d’autres tissus et organes reste encore inconnu.
Une option serait de démarrer les traitements le plus tôt possible, dans la phase présymptômatique, si possible. Dans ce cadre, un essai clinique avec le nusinersen (Etude NURTURE, ClinicalTrials.gov number, NCT02386553) est en cours. Une autre option serait de combiner les deux thérapies. Mais le coût extrêmement élevé de ce type de thérapies (750 000 Dollars pour le nusinersen) va vraisembleblement en limiter l’usage, au moins dans un premier temps.
Comme le reconnaît le Dr Van der Ploeg (Center for Lysosomal and Metabolic Diseases, Erasmus University Medical Center, Rotterdam, the Netherlands) dans the New England Journal of Medicine : « le traitement des nourrissons SMA va rapidement représenter un dilemme pour les médecins ».
Bibliographie
Cliquer sur les noms des auteurs pour accéder à la page relative à l'article sur PubMed
Date de dernière mise à jour : 16/05/2018
Ajouter un commentaire