
News sur les vaccins
Les études publiées sur les derniers résultats sont rapportées ici par ordre chronologique.
Efficacité des vaccins Pfizer et AstraZeneca sur le variant delta. 21 juillet 2021.
Lopez Bernal et al. Effectiveness of Covid-19 Vaccines against the B.1.617.2 (Delta) Variant. New England Journal of Medicine 2021.
Le variant B.1.617.2 (delta) du SARS-CoV-2 à l'origine de la Covid-19 contribue après son impact en Inde à une recrudescence des cas détectés dans le monde et notamment en France et au Royaume-Uni. Dans cette étude britannique, l'efficacité des vaccins BNT162b2 (Pfizer; vaccin à ARN) et ChAdOx1 nCoV-19 (AstraZeneca; vaccin à vecteur adénoviral) contre ce variant a été évaluée sur une cohorte de plus de 19000 personnes.
MÉTHODES
Un modèle cas-témoin (test négatif) a été utilisé pour estimer l'efficacité de la vaccination contre la maladie symptomatique causée par le variant delta ou la souche prédominante (B.1.1.7, ou variant alpha) sur la période durant laquelle le variant delta a commencé à circuler. Les variants ont été identifiés à l'aide du séquençage génomique viral et sur la base de la séquence de la protéine virale Spike (S). Les données sur tous les cas symptomatiques séquencés de Covid-19 en Angleterre ont été utilisées pour estimer la proportion de cas présentant l'un ou l'autre des variants en fonction du statut vaccinal des patients. Au total 19109 personnes ont été incluses dans l'étude, 14837 étant infectées par le variant alpha et 4272 par le variant delta.
RÉSULTATS
Après une dose de vaccin (BNT162b2 ou ChAdOx1 nCoV-19) l'efficacité était nettement plus faible chez les personnes présentant le variant delta (30,7%) que chez celles présentant le variant alpha (48,7%) ; les résultats étaient similaires pour les deux vaccins. Avec le vaccin BNT162b2, l'efficacité de deux doses était de 93,7% chez les personnes présentant le variant alpha et de 88,0 % chez celles présentant le variant delta. Avec le vaccin ChAdOx1 nCoV-19, l'efficacité de deux doses était de 74,5 % chez les personnes présentant le variant alpha et de 67,0 % chez celles présentant le variant delta.
CONCLUSIONS
Seules de modestes différences dans l'efficacité du vaccin ont été observées avec le variant delta par rapport au variant alpha après l'administration de deux doses de vaccin. Les différences absolues d'efficacité du vaccin étaient plus marquées après la réception de la première dose. Ces résultats apportent un élément objectif majeur aux efforts visant à maximiser la vaccination avec deux doses chez les populations vulnérables. (Financé par Public Health England).
Ma conclusion
Ces résultats apportent un élément objectif majeur en faveur de la vaccination dans la population générale. Ils permettent également d'expliquer pourquoi malgré la forte augmentation du nombre des cas détectés journellement au Royaume Uni (où l'infection delta est très en avance sur la France) avec 16700 cas au 24 juin 2021, puis plus de 44000 au 21 juillet, on n'assiste pas à une augmentation aussi massive du nombre des décès avec 21 décès au 24 juin, puis 102 au 21 juillet.
RAPPEL. Au 8 janvier 2021 au Royaume Uni les décès représentaient 1,9% des cas détectés (68200 cas par jour et 1300 décès par jour), alors qu'au 21 juillet 2021 les décès ne représentent que 0,2% des cas détectés (102 décès pour 44000 cas), soit approximativement 10 fois moins (données du Center for Systems Science and Engineering at Johns Hopkins University, JHU), malgré le fait que le variant delta est environ 2 fois plus infectieux que le variant alpha.
Les vaccins à ARN préviennent et atténuent la COVID-19. 30 juin 2021.
Thompson et al. Prevention and Attenuation of Covid-19 with the BNT162b2 and mRNA-1273 Vaccines. New England Journal of Medicine 2021.
Les informations sont limitées en ce qui concerne l'efficacité des vaccins à deux doses d'ARN messager (ARNm) BNT162b2 (Pfizer-BioNTech) et mRNA-1273 (Moderna) dans la prévention de l'infection par le coronavirus 2 du syndrome respiratoire aigu sévère (SARS-CoV-2) et dans l'atténuation de la maladie à coronavirus 2019 (Covid-19) lorsqu'ils sont administrés dans des conditions réelles.
MÉTHODES
Cette étude américaine issue d'un consortium de plusieurs universités, a été menée sur une cohorte prospective de 3975 membres du personnel de santé, premiers intervenants et autres travailleurs essentiels et de première ligne. Du 14 décembre 2020 au 10 avril 2021, les participants ont effectué des tests hebdomadaires de dépistage du SARS-CoV-2 sur des échantillons nasaux pharyngés mi-turbinés pour une analyse qualitative et quantitative par RT-PCR. La formule de calcul de l'efficacité du vaccin était de 100 % × (1 - rapport de risque d'infection par le SARS-CoV-2 chez les participants vaccinés par rapport aux participants non vaccinés), avec des ajustements pour tenir compte de la propension à se faire vacciner, du site de l'étude, de la profession et de la circulation virale locale.
RÉSULTATS
Le SARS-CoV-2 a été détecté chez 204 participants (5%), dont 5 seulement étaient totalement vaccinés (≥14 jours après la dose 2), 11 partiellement vaccinés (≥14 jours après la dose 1 et <14 jours après la dose 2) et 156 non vaccinés ; les 32 participants dont le statut vaccinal était indéterminé (<14 jours après la dose 1) ont été exclus. L'efficacité ajustée du vaccin était de 91% avec la vaccination complète (deux doses) et de 81% avec la vaccination partielle (une seule dose). Parmi les participants infectés, la charge moyenne d'ARN viral était inférieure de 40% chez les participants partiellement ou totalement vaccinés par rapport aux participants non vaccinés. En outre, le risque de symptômes fébriles était inférieur de 58% et la durée de la maladie était plus courte (2,3 jours de moins).
CONCLUSIONS
Les vaccins à ARNm ont été très efficaces chez les adultes en âge de travailler pour prévenir l'infection par le SARS-CoV-2 lorsqu'ils ont été administrés dans des conditions réelles, et les vaccins ont atténué la charge d'ARN viral, le risque de symptômes fébriles et la durée de la maladie chez les personnes qui ont eu une infection malgré la vaccination. (Etude financée par le National Center for Immunization and Respiratory Diseases et les Centers for Disease Control and Prevention).
Le point sur les bénéfices et effets néfastes potentiels du vaccin AstraZeneca contre la COVID-19. 7 Avril 2021.
Depuis deux semaines on parle beaucoup des cas de thromboses graves qui surviennent après la première dose du vaccin AstraZeneca. Il semble d'ailleurs que le même problème survienne avec le vaccin Johnson & Johnson nouvellement mis sur le marché. Il est donc normal que les personnes non encore vaccinées hésitent sur le choix de leur vaccin dans un contexte de disponibilité limitée des divers vaccins.
Cet article est destiné à ces personnes pour les aider à faire leur choix en toute connaissance de cause. Il est basé sur la publication du 7 Avril dernier du Winton Centre for Risk and Evidence Communication, University of Cambridge UK.
Tous les traitements médicaux (chirurgie, médicaments, vaccins) présentent des inconvénients et des avantages potentiels, et il est important de pouvoir les évaluer les uns par rapport aux autres. Dans le cas des vaccins, les avantages sont particulièrement importants car ils concernent non seulement les personnes vaccinées mais aussi celles qui ne sont pas vaccinées et qui pourraient être infectées; en revanche, les inconvénients peuvent être particulièrement dramatiques puisque les personnes qui se font vacciner sont généralement en bonne santé.
L'étude présentée ici a été réalisée au Royaume Uni, pays qui a le plus massivement vacciné avec le vaccin AstraZeneca.
Elle montre que le rapport bénéfice/risque dépend de l'âge et est largement plus élevé pour les personnes âgées de 60 ans et plus. La recommandation est donc pour les personnes les plus jeunes (âge < 55 ans) de ne pas utiliser ce vaccin (ni le Johnson & Johnson).
Contexte de l'étude
À la suite du lancement des vaccinations, une surveillance à grande échelle a été mise en place et a fait apparaître un lien potentiel entre le vaccin anti-COVID-19 d'AstraZeneca et un type spécifique de thrombose (caillot sanguin). Jusqu'au 31 mars 2021 inclus, 20,2 millions de doses du vaccin AstraZeneca ont été administrées au Royaume-Uni selon la MHRA (Medicines and Healthcare products Regulatory Agency). La MHRA a reçu 79 rapports de cas de thrombose accompagnés de faibles taux de plaquettes suite à l'utilisation du vaccin. Cela signifie que le risque global de thromboses est d'environ 4 sur 1 million de doses administrées. A noter qu'une thrombose sur 4 est mortelle (voir plus bas).
Voici les données plus détaillées tirées de ces rapports :
- 44 des 79 cas étaient des cas de thrombose veineuse cérébrale avec thrombocytopénie (diminution importante du taux de plaquettes).
- 35 des 79 cas étaient des thromboses dans d'autres veines principales avec thrombocytopénie.
- 79 cas sont survenus chez 51 femmes et 28 hommes, âgés de 18 à 79 ans. A noter que les femmes ont été plus nombreuses que les hommes à être vaccinées.
- Malheureusement, 19 personnes sont décédées (1 sur 1 million) sur les 79 cas recensés, 13 femmes et 6 hommes. Onze des 19 personnes décédées avaient moins de 50 ans, dont 3 avaient moins de 30 ans.
- Les 79 cas sont tous survenus après une première dose du vaccin.
Eléments de l'étude
Bénéfice potentiel de la vaccination
Le bénéfice potentiel de la vaccination est basé sur le nombre d'hospitalisations en service de soins intensifs évitées par le vaccin (par tranche d'âge), selon les données de l'ONS (Office of National Statistics) au 1er avril 2021 et du PHE (Public Health England) "Benefit Estimation for COVID-19" du 3 avril 2021. Une efficacité vaccinale fixe de 80% (relative à la réduction des hospitalisations en service de soins intensifs pour tous les groupes d'âge) a été utilisée dans l'étude. Les auteurs ont également choisi de considérer l'influence de trois niveaux possibles d'exposition au virus (faible, intermédiaire et élevé) en illustrant chaque fois les bénéfices accumulés sur 16 semaines. Les bénéfices du vaccin sont une protection contre la COVID-19 (COVID à court et à long terme) pour la personne vaccinée et pour les personnes avec lesquelles elles entrent en contact, car il réduit également les risques d'infection par la personne vaccinée.
Effet néfaste potentiel de la vaccination
L'effet néfaste potentiel de la vaccination est basé sur le nombre de thromboses causées par le vaccin, selon les données de la MHRA jusqu'au 31 mars par tranches d'âge. Ces taux observés ont été lissés en utilisant une régression de Poisson sur l'âge, selon une échelle logarithmique. Rappel: 1 thrombose sur 4 est mortelle. Les données de la MHRA indiquent clairement que ces cas de thrombose sont plus fréquents chez les moins de 55 ans.
Résultats
Les résultats correspondants à trois situations différentes en matière d'incidence de l'infection, faible, intermédiaire et élevée, sont présentés ci-dessous.

Situation 1. Bénéfice et effet néfaste potentiels de la vaccination avec le vaccin AstraZeneca dans le cas d'une faible exposition au virus. Dans les prévisions présentées ici, l'incidence de l'infection est de 2 sur 10 000, correspondant approximativement à la situation au Royaume-Uni en Mars 2021. A gauche (en bleu clair), les effets bénéfiques estimés de la vaccination basés sur le taux d'hospitalisations évitées en unité de soins intensifs selon les diverses tranches d'âge. On voit que l'effet bénéfique augmente avec l'âge conformément à l'observation générale selon laquelle les personnes les plus âgées sont susceptibles d'être touchées plus sévèrement que les personnes jeunes. A droite (en orange) l'incidence des effets néfastes basés sur le taux des cas de thromboses selon les diverses tranches d'âge. On voit que l'effet néfaste est plus élevé chez les personnes jeunes que chez les personnes âgées, conformément aux données de la MHRA.
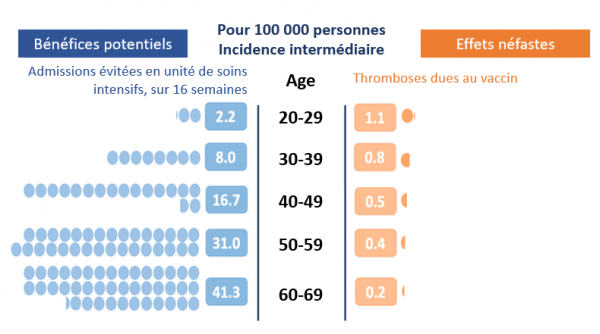
Situation 2. Bénéfice et effet néfaste potentiels de la vaccination avec le vaccin AstraZeneca dans le cas d'une exposition intermédiaire au virus. Dans les prévisions présentées ici, l'incidence de l'infection est de 6 sur 10 000, correspondant approximativement à la situation au Royaume-Uni en Février 2021. A gauche (en bleu clair), les effets bénéfiques estimés de la vaccination basés sur le taux d'hospitalisation en unité de soins intensifs selon les diverses tranches d'âge. On voit que l'effet bénéfique augmente avec l'âge conformément à l'observation générale selon laquelle les personnes les plus âgées sont susceptibles d'être touchées plus sévèrement que les personnes jeunes. Les effets bénéfiques sont supérieurs à ceux qu'ils étaient pour une incidence plus faible de l'infection (situation 1). A droite (en orange) l'incidence des effets néfastes basés sur le taux des cas de thromboses selon les diverses tranches d'âge. On voit que l'effet néfaste est plus élevé chez les personnes jeunes que chez les personnes âgées, conformément aux données de la MHRA. Par contre ces effets néfastes ne dépendent pas de l'incidence de l'infection. Pour toute tranche d'âge les effets bénéfiques potentiels augmentent par rapport aux données de la situation précédente.
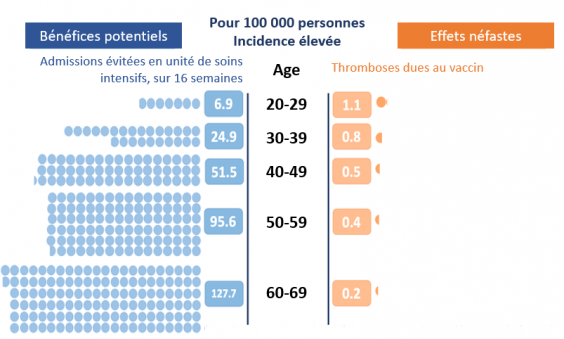
Situation 3. Bénéfice et effet néfaste potentiels de la vaccination avec le vaccin AstraZeneca dans le cas d'une forte exposition au virus. Dans les prévisions présentées ici, l'incidence de l'infection est de 20 sur 10 000, correspondant approximativement à la situation au Royaume-Uni au pic de la deuxième vague. A gauche (en bleu clair), les effets bénéfiques estimés de la vaccination basés sur le taux d'hospitalisation en unité de soins intensifs selon les diverses tranches d'âge. On voit que l'effet bénéfique augmente avec l'âge conformément à l'observation générale selon laquelle les personnes les plus âgées sont susceptibles d'être touchées plus sévèrement que les personnes jeunes. Les effets bénéfiques sont supérieurs à ceux qu'ils étaient pour une incidence plus faible de l'infection (situations 1 et 2). A droite (en orange) l'incidence des effets néfastes basés sur le nombre des cas de thrombose selon les diverses tranches d'âge. On voit que l'effet néfaste est plus élevé chez les personnes jeunes que chez les personnes âgées, conformément aux données de la MHRA. Par contre ces effets néfastes ne dépendent pas de l'incidence de l'infection. Pour toute tranche d'âge les effets bénéfiques potentiels augmentent par rapport aux données de la situation précédente.
Ces illustrations montrent le bilan approximatif tel qu'il serait pour des personnes d'âges différents, sur une période de 16 semaines, à trois expositions différentes au virus.
Dans le cas de la France, la situation 2 correspondrait approximativement à une incidence de 30 000 cas de COVID-19 par jour, ce qui est le cas aujourd'hui (16 Avril 2021). A ce jour plus de 100 000 personnes sont décédées de cette maladie en France. Depuis le 1er Mars 2021 le nombre de décès des personnes de plus de 60 ans représente 80%, alors que celui des moins de 40 ans n'est que de 5%.
Rappels
- Le risque de mort par thrombose est de 1 par million de vaccinations
- Le nombre de morts dus à la COVID est de l'ordre de 300 par jour en ce moment en France.
Pour une personne de plus de 60 ans dans un contexte d'incidence intermédiaire de l'infection (situation 2), le bénéfice de la vaccination vis-à-vis d'une hospitalisation évitée dans un service de soins intensifs est 206,5 fois plus élevé que le risque de faire une thrombose après vaccination. De plus, cette thrombose ne serait mortelle que dans un cas sur 4 (selon les données actuelles). En revanche pour une personne de moins de 30 ans, ce bénéfice n'est que deux fois plus élevé que le risque de faire une thrombose. Rappelons tout de même que pour une personne de moins de 30 ans, le risque de faire une thrombose est de 11 par million de doses administrées, le nombre des thromboses mortelles étant approximativement de 4 par million de doses administrées.
En résumé
- Chez les personnes âgées (>60 ans) le risque lié à la COVID est plus élevé, mais le risque de faire une thrombose est plus faible.
- Chez les personnes plus jeunes (<40 ans), le risque lié à la COVID est plus faible, mais le risque de faire une thrombose est plus élevé.
- Le rapport bénéfice/risque est donc beaucoup plus élevé chez les personnes âgées que chez les jeunes.
C'est la raison qui amène les Etats à conseiller le vaccin AstraZeneca uniquement pour les personnes âgées de plus de 55-60 ans.
Chez ces personnes, les bénéfices apportés par le vaccin sont beaucoup plus élevés que les risques de thrombose encourus.
Les personnes souffrant de problèmes de santé sous-jacents (comorbidités) qui augmentent le risque d'une issue défavorable de la COVID-19 bénéficieraient d'un bénéfice plus élevé du vaccin que celui illustré pour leur tranche d'âge.
Il est très important de noter que les bénéfices indiqués sont approximatifs, car ils sont pris à un niveau constant d'exposition au virus sur 16 semaines (très peu de personnes seraient susceptibles de vivre 16 semaines au taux d'exposition le plus élevé). Une personne vaccinée continuera à accumuler ces avantages pendant toute la durée de la protection offerte par le vaccin. Par contre, le risque lié à la vaccination (thrombose) ne survient qu'au moment de la vaccination. Cela signifie qu'avec le temps, les avantages augmenteront, mais pas les risques.
Il est également important de noter que les bénéfices illustrés ne concernent que l'admission en service de soins intensifs due à la COVID-19. L'étude n'illustre pas le bénéfice de ne pas propager le virus à d'autres personnes. Mais il est clair que pour chaque personne qui a été sauvée d'une admission dans un service de soins intensifs, il y en a beaucoup d'autres qui éviteront une hospitalisation et une "COVID longue" du fait de la diminution potentielle du nombre des contaminations.
Lors de la prise de décision, il est également important de prendre en compte les autres vaccins potentiels disponibles. Par exemple, s'il existait un vaccin tout aussi efficace, disponible immédiatement, et ne comportant pas le risque de thrombose, cela pourrait faire pencher la décision en faveur de ce vaccin plutôt que celui d'AstraZeneca ou de J&J. Cependant, si un tel vaccin n'était pas immédiatement disponible, le risque d'exposition au virus SARS-CoV-2 pendant chaque semaine de retard avant qu'un tel vaccin ne soit disponible devrait être pris en compte dans la décision d'attendre ou non.
Tous ces facteurs font que toute décision concernant le vaccin d'AstraZeneca est complexe, le rapport risque/bénéfice variant selon les personnes et selon la prévalence du virus.
Au final
Il est particulièrement regrettable que le vaccin AstraZeneca soit responsable de thromboses sévères, car le laboratoire s'était engagé (et a tenu ses promesses) à faire payer le vaccin à prix coûtant, soit 1,78€ par dose. Le prix de la dose pour les deux autres vaccins sur le marché en France actuellement (Pfizer/BioNtech et Moderna) est de 15€.
_____________________________________________________________________________________________________________________________________________
Réaction des experts aux informations selon lesquelles le Danemark a suspendu la vaccination avec le vaccin AstraZeneca par mesure de précaution après certains rapports de thrombose. Science Media Center, 11 MARS 2021.
Il a été rapporté que la vaccination avec le vaccin AstraZeneca anti-COVID-19 est suspendue jusqu'à nouvel ordre au Danemark après des rapports de cas de thrombose.
Divers experts britanniques répondent sur ce sujet.
Selon ces experts, la suspension d'utilisation du vaccin AstraZeneca n'est pas justifiée pour les raisons suivantes:
- Selon les données disponibles dans la population générale (sans vaccination récente), le taux de thrombose est de 1 à 2 cas sur 1000 mais augmente avec l'âge. Ce taux est très supérieur à ce qui est observé après la vaccination avec le vaccin AstraZeneca, soient 22 cas sur 3 milions (7 cas par million) de personnes vaccinées, identifiés par l'Agence Européenne du Médicament.
- Au Royaume Uni, plus de 11 millions de doses du vaccin AstraZeneca ont été administrées au cours des derniers mois, et aucun excès du nombre des thromboses n'a été signalé parmi les personnes recevant le vaccin, par rapport au nombre attendu dans la population générale.
- Une infection grave de COVID-19 elle-même peut provoquer des thromboembolies par un dysfonctionnement du système immunitaire.
- Il n'a pas été confirmé scientifiquement que l'observation d'une thromboembolie au Danemark (ou ailleurs) ait été causée par le vaccin COVID-19 AstraZeneca.
Conclusion des experts
S'il existe un lien entre le vaccin et la coagulation du sang, le risque est vraisemblablement très faible.
Avis des experts (traduits en Français par PM)
Professeur Paul Hunter, professeur de médecine à la Norwich School of Medicine de l'Université d'East Anglia
Il y a eu un petit nombre de rapports de thromboembolie (formation de caillots sanguins dans les vaisseaux sanguins puis blocage de la circulation sanguine) suite à l'administration du vaccin AstaZeneca possiblement associé à un seul lot de vaccin.
C'est une pratique courante, en particulier avec tout nouveau médicament ou vaccin, que de signaler tout événement indésirable survenant après l'administration. Au Royaume-Uni, cette pratique est connue sous le terme de programme du "carton jaune". Ces rapports sont établis que le médecin déclarant sache ou non que l'événement a été causé par un médicament ou un vaccin. En effet, chez un patient donné, il est souvent impossible de savoir si un événement indésirable a été causé par le vaccin ou s'il se serait produit de toute façon. Ceci est particulièrement difficile lorsque les personnes recevant le vaccin sont susceptibles de souffrir de diverses pathologies, notamment comme les personnes âgées. C'est certainement le cas pour les personnes qui ont bénéficié des premières vagues de vaccination.
Étant donné le grand nombre de types différents d'événements indésirables possibles qui pourraient être signalés, il est très probable que certains événements indésirables soient identifiés lors d'une campagne de vaccination comme celle-ci, même lorsque le vaccin ne joue aucun rôle dans la causalité de l'événement indésirable. Ainsi, bien que la plupart de ces troubles thromboemboliques n'aient pas été causés par le vaccin, cela ne signifie pas qu'ils doivent être ignorés. Ils doivent être correctement étudiés.
L'une des questions à prendre en compte est la fréquence de la thromboembolie. Un article récent sur cette maladie a étudié son incidence en 2012, 2013 et 2014 aux Etats Unis (Wendelboe et al. Incidence of Venous Thromboembolism in a Racially Diverse Population Oklahoma County, Oklahoma. Thrombosis and Hemostasis 2021). Le principal facteur affectant l'incidence de la thromboembolie dans la communauté était l'âge. L'incidence de la thromboembolie veineuse était de 13,16 dans le groupe d'âge> 80 ans, de 7,76 dans le groupe d'âge 70-79 ans et de 5,10 dans le groupe d'âge 60-69 ans pour 1000. Donc, pour chaque million de personnes vaccinées dans ces groupes d'âge, nous nous attendons à voir de toute façon environ 1097, 645 et 425 épisodes de thromboembolie dans le mois suivant la vaccination.
Avec les informations actuellement disponibles, il semble probable que ces événements indésirables signalés ne soient pas causés par le vaccin, mais nous verrons à quelles conclusions les autorités compétentes en arriveront après avoir examiné toutes les preuves.
Professeur Adam Finn, professeur de pédiatrie à l'Université de Bristol
Quand quelque chose de grave se produit après avoir été vacciné, il est assez naturel de se demander si le vaccin en était la cause. Cependant, lorsqu'un très grand nombre de personnes sont vaccinées sur une courte période, un certain nombre de maladies inattendues et inhabituelles vont survenir dans la période suivant la vaccination du fait du hasard.
Si un nombre suffisant d'évènements émerge, la suspicion qu'il pourrait y avoir un lien de causalité avec le vaccin augmente, surtout s'il y a un mécanisme qui pourrait expliquer comment les deux événements pourraient conduire de l'un à l'autre.
La position avec le vaccin AstraZeneca pour le moment est qu'il n'y a aucun signe, nulle part, y compris au Royaume-Uni où un très grand nombre de doses ont maintenant été administrées, que les maladies liées aux thromboses se produisent plus fréquemment que d'habitude.
C'est plutôt rassurant car cela signifie soit que le vaccin ne provoque pas du tout de thrombose, soit, au pire, que c'est un événement extrêmement rare.
Il est vraiment important de surveiller l'innocuité de tout vaccin en permanence surtout s'il est largement utilisé. Les autorités britanniques communiqueront avec celles des autres pays et continueront à assurer leur propre surveillance.
En attendant, le rapport bénéfice / risque est très clair. Mieux vaut se faire vacciner dès que c'est possible, à la fois pour minimiser votre risque personnel et pour aider à maîtriser l'épidémie afin que nous puissions tous progresser vers un avenir plus normal.
Professeur Jon Gibbins, directeur de l'Institut de recherche cardiovasculaire et métabolique, Université de Reading
Je pense qu'il est tout à fait compréhensible que les gens puissent s'inquiéter à ce sujet, mais plusieurs facteurs nous obligent à être prudents quant au lien entre le vaccin et la coagulation.
La coagulation du sang, ou thrombose, survient pour diverses raisons, et peut souvent ne pas être détectée dans la population générale où la thrombose veineuse est relativement courante et affecte 1 à 2 personnes sur 1000. Les raisons qui ont poussé le Danemark à faire une pause l'utilisation du vaccin AstraZeneca ne sont pas claires, mais je note que l'EMA (Agence Européenne du Médicament) a identifié 22 cas de thrombose parmi les 3 millions qui ont reçu le vaccin jusqu'à présent, ce qui est bien inférieur à la prévalence de 1 à 2 sur 1000 pour la thrombose. Par conséquent, s'il existe une association entre le vaccin et la coagulation, le risque est vraisemblablement très faible.
Il est important de noter qu'il existe de bonnes preuves émergentes selon lesquelles une infection grave de COVID-19 elle-même peut provoquer des thromboembolies par un dysfonctionnement du système immunitaire, et il se peut donc que les gens soient plus à risque de coagulation parce qu'ils ne sont pas immunisés. Nous aurons besoin de voir les données détaillées afin d'évaluer cela correctement, mais à ce stade, nous devons également faire attention à ne pas provoquer de panique ou de résistance injustifiée à la vaccination.
Docteur Andrew Garrett, vice-président exécutif, Opérations scientifiques, ICON Clinical Research
D'après ce que nous avons vu des données publiées jusqu'à présent, les vaccins contre le coronavirus sont très sûrs. Bien entendu, à mesure que les vaccins sont largement déployés via des programmes de vaccination nationaux, les événements indésirables seront observés à des fréquences qui reflètent l'ampleur du déploiement. La nature même de la priorisation des vaccins signifie également que de nombreux événements observés seront confondus avec les comorbidités associées aux personnes âgées et aux personnes cliniquement vulnérables. Il est beaucoup plus difficile de déterminer la causalité avec des données d'observation en raison de l'absence de contrôle simultané et de randomisation, tandis que démêler les différents effets potentiels peut être un défi. C'est pourquoi les régulateurs tels que l'EMA ont un plan de pharmacovigilance pour les vaccins autorisés.
Les essais cliniques de vaccins eux-mêmes étaient très importants - beaucoup plus importants que pour la plupart des programmes de développement clinique de traitements - ils avaient donc en fait une chance beaucoup plus élevée que d'habitude de détecter des événements indésirables très rares tout en caractérisant très bien les événements courants (comme les réactions au site d'injection). Par exemple, les chances d'observer au moins un événement très rare «1 sur 10 000» sont de 78% avec 15 000 participants à un essai clinique recevant un vaccin expérimental. Il y a donc de bonnes chances d'observer des événements rares et très rares avant l'approbation d'un vaccin, et les essais cliniques ont l'avantage d'un groupe témoin pour comparer objectivement la fréquence des événements. En outre, les données de sécurité de ces essais cliniques rapportés s'accumulent encore puisque les participants à ces études sont suivis pendant deux ans.
Une fois qu'un vaccin (ou tout médicament) est déployé, l'approche de surveillance standard consiste à examiner les événements excessifs d'un type particulier dans un processus connu sous le nom de détection de signal. Ces événements peuvent être signalés à partir de diverses sources et, en Europe, sont stockés dans le système EudraVigilance. Les événements excessifs, par exemple, peuvent être estimés en comparant les événements enregistrés pendant le déploiement du vaccin avec ceux provenant de grandes bases de données de taux d'événements sous-jacents, y compris pour des produits similaires. Ces bases de données sont maintenues et mises à jour par des groupes spécifiques et incluent la base de données Sentinel de la FDA qui utilise des données électroniques sur les soins de santé et les réclamations administratives. Des types particuliers d'événements peuvent également être examinés plus en détail grâce à des comparaisons de sous-groupes (comme le sexe, l'âge, etc.). Il est également possible de réexaminer les données des essais cliniques à la recherche de signes spécifiques d'un événement qui a été observé lors du déploiement. Par exemple en réexaminant des échantillons de sang.
Il est inévitable que des événements graves soient observés lors de la vaccination contre le coronavirus, comme ils l'étaient avant le programme de vaccination. Il incombe aux promoteurs et aux agences de réglementation de surveiller attentivement ces événements et d'évaluer si la fréquence des événements observés est disproportionnée par rapport à ce qui aurait été normalement prévu et, dans l'affirmative, d'en déterminer la cause potentielle. L'Agence danoise des médicaments a adopté une approche prudente suite à l'observation de thromboses et d'un décès, et poursuit ses investigations tout en interrompant la vaccination pendant 14 jours. Ce faisant, ils ont dû trouver un équilibre entre les avantages et les risques d'un éventuel retard de vaccination au Danemark. »
Professeur Anthony Harnden, vice-président du Comité mixte sur la vaccination et l'immunisation
La sécurité des vaccins est d'une importance cruciale. Notre organisme de réglementation britannique, la MHRA (Medicines and Healthcare products Regulatory Agency), examine tous les rapports d'événements indésirables pour les vaccins au fur et à mesure qu'ils sont signalés. Il y a eu plus de 11 millions de doses du vaccin AstraZeneca administrées au Royaume-Uni et aucun excès de rapports de thromboses parmi les personnes recevant le vaccin, par rapport au taux attendu dans la population. Le public doit avoir la certitude que les vaccins utilisés dans le programme de vaccination britannique sont sûrs et hautement efficaces pour prévenir les maladies graves, y compris la prévention des thromboses causées par la COVID-19.
Docteur Peter English, consultant à la retraite en contrôle des maladies transmissibles, ancien rédacteur en chef du magazine Vaccines in Practice, ancien président du comité de médecine de santé publique BMA (British Medical Association)
Il n'est en aucun cas inhabituel que l'introduction d'un nouveau vaccin soit interrompue par des rapports d'événements indésirables comme celui-ci. C'est un signe que les systèmes de surveillance des effets indésirables fonctionnent - mais généralement pas un signe que les effets indésirables sont causés par la vaccination.
Lorsque des vaccins sont administrés à des millions de personnes, il faut seulement admettre que des troubles médicaux arriveront à certaines d'entre elles après avoir été vaccinées. Ce qui est important, c'est de pouvoir évaluer si ces «événements indésirables» sont liés de manière causale au vaccin.
À cette fin, il est important d'entreprendre des évaluations sur la plausibilité biologique et, plus important encore, des calculs pour savoir si le nombre de ces événements est supérieur ou inférieur à ce qui serait attendu par hasard si les personnes n'avaient pas été vaccinées.
Avec des résultats rares ou inhabituels, il peut être particulièrement difficile de déterminer si les événements sont plus fréquents après la vaccination. Des organisations telles que la Brighton Colaboration coordonnent certains de ces travaux; et en Angleterre (comme ailleurs), le Public Health England a une stratégie rigoureuse de surveillance des vaccins.
Cela peut être particulièrement difficile lorsque la maladie associée (et peut-être mais pas nécessairement causée par) la vaccination est une séquelle établie de la maladie que le vaccin est utilisé pour prévenir. Avec le syndrome de Guillain-Barré et la vaccination contre la grippe, par exemple, le nombre de cas après vaccination est si faible qu'il est difficile de savoir s'il y a ou non une augmentation par rapport aux taux de base; en revanche, la grippe cause définitivement le syndrome de Guillain-Barré. Donc, pour éviter le syndrome de Guillain-Barré, étant donné qu'il est difficile d'éviter l'exposition à la grippe, l'option la plus sûre est de se faire vacciner (car la vaccination réduit votre risque de contracter la grippe et donc de contracter le syndrome de Guillain-Barré causé par la grippe; et cet effet l'emporte largement sur tout risque minime lié à la vaccination).
Les experts ne savent toujours pas si les événements rapportés au Danemark - et, en fait, le décès signalé à Vienne - sont liés de manière causale au vaccin. Je suppose qu’il s’agit plus souvent d’associations fortuites que de liens de causalité.
Docteur Phil Bryan, responsable de la sécurité des vaccins de la MHRA
La sécurité des vaccins est d'une importance capitale et nous surveillons en permanence la sécurité des vaccins pour nous assurer que les avantages l'emportent sur les risques potentiels. Il n'a pas été confirmé scientifiquement que l'observation d'une thromboembolie au Danemark ait été causée par le vaccin COVID-19 AstraZeneca.
L'action des autorités danoises pour suspendre temporairement l'utilisation du vaccin est une mesure du principe de précaution pendant qu'elles enquêtent.
Les thromboses peuvent se produire naturellement et ne sont pas rares. Plus de 11 millions de doses du vaccin COVID-19 AstraZeneca ont maintenant été administrées à travers le Royaume-Uni. Les rapports de thrombose reçus jusqu'à présent ne sont pas supérieurs au nombre qui se serait produit naturellement dans la population non vaccinée.
La sécurité du public passera toujours en premier. Nous suivons de près cette question, mais les preuves disponibles ne confirment pas que le vaccin en est la cause.
Les gens doivent continuer à se faire vacciner contre le COVID-19. La MHRA encourage quiconque à signaler tout soupçon ou préoccupation au-delà des effets secondaires bénins connus sur le site Coronavirus Yellow Card. Les déclarants n'ont pas besoin d'être sûrs d'un lien entre un vaccin et un effet secondaire suspecté, mais sont tout de même encouragés à le signaler.
Professeur Stephen Evans, professeur de pharmacoépidémiologie à la London School of Hygiene & Tropical Medicine
Il s'agit d'une approche extrêmement prudente basée sur certains rapports isolés en Europe (Danemark et Autriche, notamment). Le problème avec les notifications spontanées de réactions indésirables suspectées après une vaccination est l'énorme difficulté de distinguer un effet causal d'une coïncidence.
Cela est particulièrement vrai lorsque nous savons que la maladie COVID-19 est très fortement associée à la coagulation sanguine et qu'il y a eu des centaines, voire plusieurs milliers de décès dus à la coagulation sanguine à la suite de la maladie de COVID-19. La première chose à faire est d'être absolument certain que les thromboses n'avaient pas d'autre cause, y compris la COVID-19.
Une approche rationnelle consiste à enquêter et à s'assurer que le rapport bénéfice / risque est en faveur du vaccin. Étant donné que nous savons avec une grande certitude que le vaccin prévient la COVID-19 et que nous sommes presque totalement certains que le vaccin n'ait pu causer ce problème, le rapport bénéfice / risque est toujours très favorable au vaccin à mon avis.
Si toutefois, il n'y a aucune pénurie de vaccins alternatifs, alors une approche de précaution extrême telle qu'elle est adoptée au Danemark peut être justifiée. Toutefois, si cette approche empêche certaines personnes de se faire vacciner alors qu'elles sont vulnérables à la COVID-19, alors c'est un principe de précaution erroné.
Pour autant que l'on sache, il n'y a pas eu de « signal » de tels problèmes au Royaume-Uni et même s'il y avait un « signal », basé sur des rapports spontanés, une étude épidémiologique appropriée et rapide serait nécessaire pour voir s'il s'agit ou non d'une coïncidence.
Déclarations de conflits d'intérêt
Prof Paul Hunter. Le seul conflit d'intérêt est que j'ai été vacciné mardi et que c'était le vaccin AZ.
Prof Adam Finn est un chercheur impliqué dans les essais cliniques du vaccin AstraZeneca et d'autres vaccins anti-COVID-19. Il est membre du JCVI (Joint Committee on Vaccination and Immunisation), membre du groupe de travail OMS et SAGE (Organisation Mondiale de la Santé et Groupe stratégique consultatif d'experts) sur les vaccins anti-COVID-19 et président du groupe consultatif technique européen d'experts de la vaccination de l'OMS.
Prof Jon Gibbins est financé par la British Heart Foundation pour ses recherches sur la thrombose, y compris des travaux sur la coagulation sanguine causée par COVID-19 dans les poumons.
Dr Andrew Garrett est employé par ICON, une organisation de recherche sous contrat qui fournit des services pharmaceutiques aux industries pharmaceutique et biotechnologique. ICON mène des essais cliniques pour le compte des sponsors, y compris des essais de vaccins. Andrew est membre du panel d'accréditation de recherche de l'Autorité statistique britannique (UKSA).
Dr Peter English : pas de conflit d'intérêt.
Prof Stephen Evans : aucun conflit d'intérêt. Financé (un jour par semaine) par la LSHTM (London School of Hygiene & Tropical Medicine) qui reçoit des financements de diverses entreprises, dont AstraZeneca et GSK (Glaxo Smith Klein), mais non financé par eux ces entreprises, non impliqué dans l'obtention de financement de leur part et non chercheur sur les subventions obtenues de leur part. Est le statisticien du «Meta-Data Safety and Monitoring Board» du CEPI (Coalition for Epidemic Preparedness Innovations). Est payé pour sa participation à ces réunions. Participe à l'essai Oxford/Astra Zeneca et le 13 janvier 2021, a appris avoir reçu le vaccin AZ actif.
Voir le document original (en Anglais) ICI
________________________________________________________________________________________________________
Vaccins Pfizer et AstraZeneca : résultats comparatifs sur la vaccination en Ecosse (Février 2021)
Vasileiou et al. Effectiveness of first dose of COVID-19 vaccines against hospital admissions in Scotland: national prospective cohort study of 5.4 million people. SSRN Lancet preprint 2021
L'étude présentée ici a été conduite par des médecins et chercheurs opérant dans diverses universités Ecossaises ou dans divers services de Santé Publique Ecossais.
Méthodes
Une cohorte d'observation prospective ouverte, en temps réel, avec une couverture nationale en Écosse a été construite en utilisant un ensemble d'informations cliniques unique composé de données liées à la vaccination, aux soins primaires, aux tests de laboratoire (RT-PCR et signes cliniques de la COVID-19), aux hospitalisations et à la mortalité (dues à une COVID avérée). Les données étaient disponibles pour 5,4 millions de personnes.
L'étude comparative a porté sur l'effet de la première dose des vaccins Pfizer (BNT162b2 mRNA COVID-19) et AstraZeneca (ChAdOx1 nCoV-19) administrée entre le 8 décembre 2020 et le 15 février 2021. Les groupes de personnes vaccinées ont été stratifiés par intervalles de temps de 7-13, 14-20, 21-27, 28-34, 35-41 et >42 jours après la vaccination, par type de vaccin administré (Pfizer ou AstraZeneca), et par groupes d'âge (18-64 ans, 65-79 ans et >80 ans).
L'efficacité des vaccins a été évaluée en prenant en compte dans les groupes vaccinés et non vaccinés, le nombre des hospitalisations pour cause de COVID-19 (confirmée par données RT-PCR et données cliniques), ou les admissions hospitalières dans les 28 jours suivant un test RT-PCR positif, du 8 décembre 2020 au 13 février 2021. L'efficacité a ainsi été évaluée par le taux de dangerosité (HR, Hazard Ratio). Le taux de dangerosité est un paramètre statistique mesurant le risque de survenue d'un événement (ici hospitalisation pour cause de COVID-19) dans un groupe (vaccinés) par rapport à l’autre (non vaccinés). Par exemple, si la population vaccinée a un risque d'être hospitalisée 4 fois moins élevé que celui de la population non vaccinée, le taux de dangerosité est de 0,25 pour la population vaccinée. On en conclut que l'efficacité du vaccin est de 75%.
Le premier critère d'évaluation portait sur l'efficacité de chacun des deux vaccins; le deuxième portait sur l'efficacité du statut vaccinal global (les deux vaccins) stratifiée par groupes d'âge (18-64 ans, 65-79 ans et >80 ans).
Résultats
Entre le 8 décembre 2020 et le 15 février 2021, 1 137 775 (35 %) patients ont été vaccinés. Une acceptation rapide des vaccins Pfizer et AstraZeneca a été observée au cours de la période d'étude. Pour le vaccin Pfizer, le taux d'utilisation le plus élevé a été constaté chez les patients <65 ans, tandis que pour le vaccin AstraZeneca une plus grande utilisation du vaccin a été constatée chez les patients >80 ans.
Les résultats présentés ci-dessous montrent que le vaccin AstraZeneca est aussi efficace que le vaccin Pfizer en termes de réduction du nombre des hospitalisations liées à une COVID-19 avérée. Ils montrent également que leur efficacité est maximale environ un mois après administration de la première dose.
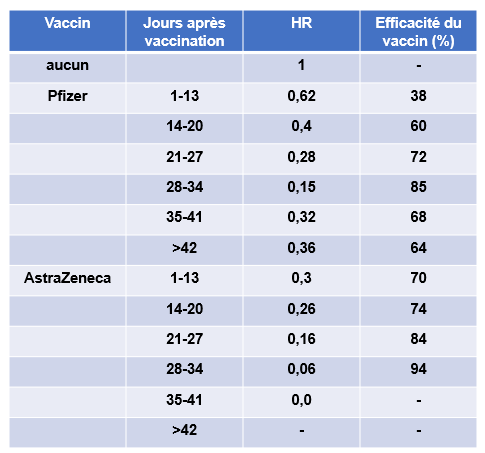
Efficacité des vaccins Pfizer et AstraZeneca en fonction du nombre de jours après administration (un dose)
HR: taux de dangerosité (Hazard Ratio) estimé à partir du nombre des hospitalisations dues à la COVID-19 pendant l’étude.
Adapté de Vasileiou et al. SSRN Lancet 2021 (preprint).
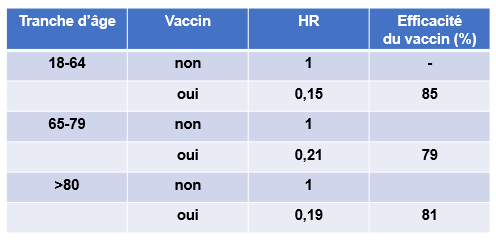
Efficacité de la vaccination (Pfizer et AstraZeneca) en fonction de la tranche d’âge
HR: taux de dangerosité (Hazard Ratio) estimé à partir du nombre des hospitalisations dues à la COVID-19 pendant l’étude.
Adapté de Vasileiou et al. SSRN Lancet 2021 (preprint).
Cette étude de cohorte prospective nationale comprenant la quasi-totalité de la population écossaise a démontré qu'une dose unique des vaccins Pfizer et AstraZeneca était associée à une protection substantielle contre les hospitalisations liées à une COVID-19, avec des efficacités maximales de 85% pour le vaccin Pfizer et de 94% pour le vaccin AstraZeneca. Dans le groupe d'âge le plus élevé (≥80 ans), sur la base d'une analyse groupée pour les deux vaccins, une efficacité maximale de 81% a été observée.
Conclusion
Cette étude apporte des preuves convaincantes que la première dose des vaccins Pfizer et AstraZeneca protège contre les hospitalisations dues à une COVID-19 chez les adultes et les personnes de plus de 80 ans. La période de protection maximale apparaît entre 28 à 35 jours après administration du vaccin.
Phase 3 du vaccin Janssen - Johnson & Johnson. Les résultats présentés ici sont issus du document fourni par Janssen et Johnson & Johnson à la FDA (26 Février 2021) (https://www.fda.gov/media/146217/download).
Janssen et Johnson & Johnson ont développé un vaccin à vecteur viral constitué d'un adénovirus de sérotype 26 recombinant pour la protéine S pleine longueur et stabilisée du SARS-CoV-2, appelé Ad26.COV2.S.
Un essai clinique de phase 3 randomisé, en double aveugle et contrôlé par placebo appelé "ENSEMBLE" a été mis en place en septembre 2020 et s'est terminé le 17 décembre 2020. Son but était d'évaluer l'innocuité et l'efficacité du vaccin à dose unique chez des volontaires adultes âgés de 18 ans et plus. Les participants à l'étude ont reçu une seule injection intramusculaire d'Ad26.COV2.S à un niveau de dose de 5 × 1010 particules virales le premier jour. L'essai a été suspendu provisoirement entre les 12 et 23 octobre 2020 après observation d'un problème grave chez un volontaire. L'essai a repris le 23 Octobre après que l'analyse des données médicales ait écarté le vaccin comme source de ce problème.
L'analyse intermédiaire d'efficacité (à la date du 22 janvier 2021) a inclus 39 321 participants avec un temps de suivi médian de 2 mois après vaccination. L'efficacité du vaccin contre les cas modérées à sévères/critiques de COVID-19 confirmées en laboratoire par RT-PCR dans toutes les zones géographiques où l'essai a été mené était de 66,9 % si l'on considère les cas survenus au moins 14 jours après vaccination et de 66,1 % si l'on considère les cas survenus au moins 28 jours après la vaccination. Pour les groupes vaccin et placebo, respectivement, 116 et 348 cas de COVID-19 sont survenus au moins 14 jours après la vaccination, et 66 et 193 cas sont survenus au moins 28 jours après la vaccination.
L'efficacité du vaccin contre les cas graves/critiques de COVID-19 confirmés en laboratoire par RT-PCR survenus au moins 14 jours et au moins 28 jours après la vaccination, était de 76,7% et 85,4, respectivement. Dans une analyse post hoc de toutes les hospitalisations liées à la COVID-19 commençant 14 jours après la vaccination, il y avait 2 cas dans le groupe vaccin (avec aucun cas après 28 jours) contre 29 cas dans le groupe placebo (avec 16 cas après 28 jours). Au 5 février 2021, il y avait 7 décès liés à la COVID-19 dans le groupe placebo et aucun dans le groupe vacciné.
En général, l'efficacité dans les sous-groupes (âge, comorbidité, race, ethnicité) semble être similaire à l'efficacité dans l'ensemble de la population étudiée. Une estimation plus faible de l'efficacité a été observée pour le sous-groupe des participants de 60 ans et plus présentant des comorbidités par rapport à l'ensemble de la population, mais avec une tendance observée à l'augmentation de l'efficacité à mesure que le nombre de cas inclus dans l'analyse augmentait (c'est-à-dire en comptant les cas à partir de 14 jours plutôt que 28 jours et en incluant les cas non encore confirmés de manière centralisée). Il n'y a eu aucun décès lié à la COVID-19 et aucun cas de COVID-19 nécessitant une intervention médicale survenu 28 jours ou plus après la vaccination chez les participants âgés de 60 ans ou plus présentant des comorbidités médicales dans le groupe vacciné.
Les souches prédominantes parmi celles qui ont été séquencées à partir des prélèvements réalisés sur les participants étaient la variante Wuhan-H1 (D614G) aux États-Unis (96,4 % des cas séquencés), la variante 20H/501Y.V2 (B.1.351) en Afrique du Sud (94,5 % des cas séquencés) et la variante de la lignée P.2 (apparenté au variant B.1.1.28 P1) au Brésil (69,4 % des cas séquencés, les 30,6 % restants étant la variante Wuhan-H1 D614G). Aucun cas n'a été identifié comme étant de la lignée B.1.1.7 ou B.1.1.28 P1 au 12 février 2021. Voir sur ce site la page "Les variants du SARS-CoV-2".
L'analyse de sécurité du vaccin jusqu'à la date limite du 22 janvier 2021 a porté sur 43 783 participants avec un suivi médian de 2 mois. L'analyse a confirmé un profil de sécurité favorable, sans qu'aucun problème de sécurité spécifique n'ait été identifié. Les effets indésirables sollicités les plus fréquents associés à l'Ad26.COV2.S étaient la douleur au point d'injection (48,6 %), les céphalées (38,9 %), la fatigue (38,2 %) et la myalgie (33,2 %) ; ils étaient principalement légers et modérés, 0,7 % et 1,8 % des effets indésirables locaux et systémiques sollicités, respectivement, étant de grade 3.
L'intérêt de ce vaccin est qu'il ne nécessite qu'une seule dose. Il est par ailleurs stable à -20°C pendant plusieurs mois.
En février 2021, Sanofi et Johnson & Johnson ont conclu un accord pour que le groupe Français assure un soutien et une infrastructure dans son usine de Marcy-l'Étoile pour la fabrication d'environ 12 millions de doses de vaccin par mois dès que l'autorisation de l'ANSM aura été reçue (Mars-Avril 2021).
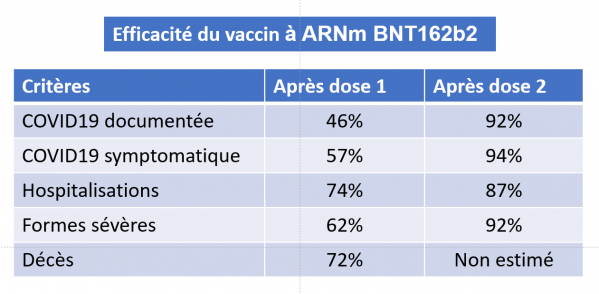
Vaccination en Israël: des résultats très encourageants (24 Février 2021)
Dagan et al. BNT162b2 mRNA Covid-19 Vaccine in a Nationwide Mass Vaccination Setting. New England Journal of Medicine 2021
Alors que les campagnes de vaccination de masse contre la COVID-19 se mettent en place dans le monde entier, l'efficacité des vaccins doit être évaluée par toute une série de résultats sur des populations diverses dans un cadre non contrôlé. Dans cette étude, les données de la plus grande organisation de soins de santé d'Israël (Clalit Health Services) ont été utilisées pour évaluer l'efficacité du vaccin à ARNm BNT162b2 (Pfizer-BioNtech). Il s'agit à proprement parler d'une étude de phase 4, c'est à dire une étude permettant de valider l'efficacité du vaccin dans la "vraie vie", faisant suite aux résultats obtenus dans l'essai de phase 3 (voir plus bas sur ce site, l'article de Polak et al. 31 Décembre 2020). Plusieurs articles avaient déjà été publiés sur le sujet sur la plateforme MedRxiv 2021 (Chodick et al.; Rossman et al.; Levine-Tiefenbrun et al.).
Méthodes
Toutes les personnes qui ont été nouvellement vaccinées entre le 20 décembre 2020 et le 1er février 2021 (près de 600 000 personnes enregistrées auprès de l'organisation de soins Clalit Health Services) ont été comparées à des témoins non vaccinés dans un rapport de 1:1 en fonction de leurs caractéristiques démographiques et cliniques.
Les critères principaux de jugement de cette étude étaient les suivants: i) apparition d'infections par le SARS-CoV-2 confirmées par RT-PCR, ii) COVID-19 symptomatiques, iii) hospitalisations liées à la COVID-19, iv) COVID-19 graves et v) décès. L'analyse statistique des résultats s'est faite selon l'estimateur de Kaplan-Meier.
Résultats
Chaque groupe d'étude comprenait 596 618 personnes. Au cours d’un suivi médian de 15 jours, 10 561 infections dues au SARS-CoV-2 (0,6 pour 1 000 sujets-jours) confirmées par RT-PCR ont été dénombrés dont 5 996 (57 %) ont conduit à des formes symptomatiques. Une hospitalisation a été nécessaire chez 369 patients et dans 229 cas, il s’agissait d’une forme sévère de la maladie ; le nombre de décès s’est élevé à 41.
L'efficacité estimée du vaccin, égale à : 1- (taux d'attaque chez vaccinés/taux d'attaque chez non-vaccinés) x 100, entre les jours 14 à 20 après la première dose et à 7 jours ou plus après la deuxième dose était la suivante : i) pour une infection confirmée, 46% (95% de confiance 40 à 51) et 92 % (IC à 95 %, 88 à 95) ; pour une COVID-19 symptomatique, 57 %. (IC 95%, 50 à 63 ans) et 94% (IC 95%, 87 à 98 ans) ; pour une hospitalisation, 74% (IC 95%), 56 à 86) et 87% (IC 95%, 55 à 100) ; et pour les formes graves, 62% (IC 95%, 39 à 80) et 92 % (95 % IC, 75 à 100), respectivement. L'efficacité estimée de la prévention des décès dus à la COVID-19 était de 72 % (IC à 95 %, 19 à 100) pour les jours 14 à 20 après la première dose.
L’efficacité vaccinale s’est avérée identique dans toutes les tranches d’âge et légèrement plus faible en cas de comorbidités multiples (> ou = 3).
Deux facteurs ont fait que la présente étude est particulièrement adaptée à l'évaluation de l'efficacité du vaccin BNT162b2 dans une application pratique :
1. Une combinaison rare de données médicales de base, résultats de tests PCR, données de suivi des patients dans la communauté et dans les établissements hospitaliers grâce au travail du Clalit Health Services qui a maintenu ces données intégrées pour plus de la moitié de la population israélienne, et l'a mis à jour quotidiennement, pendant plus de deux décennies.
2. Le rythme rapide et la forte absorption du vaccin Covid-19 en Israël et les taux élevés de maladie pendant la campagne de vaccination.
Conclusions
Cette étude, réalisée dans le cadre d'une vaccination de masse à l'échelle nationale, suggère que le vaccin à ARNm BNT162b2 est efficace pour un large éventail de manifestations cliniques liés à la COVID-19. Les résultats de l'étude sont conformes à ceux publiés concernant la phase 3 de l'essai randomisé.
Sources et financements de cette étude
Clalit Research Institute, Innovation Division, Clalit Health Services, Tel Aviv Israël; School of Public Health, Faculty of Health Sciences, Ben Gurion University of the Negev, Be’er Sheva Israël; University of Michigan School of Public Health, Ann Arbor USA; Department of Biomedical Informatics, Harvard Medical School; Departments of Epidemiology and Biostatistics, Center for Communicable Disease Dynamics, Departments of Epidemiology and of Immunology and Infectious Diseases, Harvard T.H. Chan School of Public Health, Harvard–MIT Division of Health Sciences and Technology, et Predictive Medicine Group, Computational Health Informatics Program, Boston Children’s Hospital, Boston USA.
Note supplémentaire
Selon une étude (Munitz et al. MedRxiv 2021) portant sur plus de 300 000 échantillons analysés par RT-PCR recueillis entre le 6 décembre 2020 et le 10 février 2021 (une période couvrant l'étude de Dagan et al. résumée ci-dessus) dans la communauté générale et les maisons de retraite, il a été observé qu'en l'espace de six semaines, le variant B.1.1.7 (Britannique) était capable de concurrencer la souche sauvage du SARS-CoV-2 pour devenir le variant principal en Israël. La transmission du variant B.1.1.7 dans la population des plus de 60 ans a pratiquement cessé, montrant ainsi l'efficacité de vaccin vis à vis de ce variant.
Vaccin Sputnik, essai de phase 3 (2 Février 2021)
Logunov et al. Safety and efficacy of an rAd26 and rAd5 vector-based heterologous prime-boost COVID-19 vaccine: an interim analysis of a randomised controlled phase 3 trial in Russia. Lancet 2021.
Cet article présente les résultats préliminaires sur l'efficacité et la sécurité du vaccin Sputnik V (Gam-COVID-Vac), issus de l'analyse intermédiaire de l'essai de phase 3. Sputnik V est un vaccin hétérologue recombinant à base de vecteur viral, deux adénovirus rAd26 et rAd5, les deux vecteurs étant porteurs du gène de la glycoprotéine Skipe (S) pleine longueur du SRAS-CoV-2.
Méthodes
Un essai de phase 3 randomisé, en double aveugle et contrôlé par placebo a été réalisé dans 25 hôpitaux et polycliniques de Moscou, en Russie. Les participants étaient âgés d'au moins 18 ans, avec des résultats négatifs aux tests PCR et sérologiques (IgG et IgM) du SRAS-CoV-2; ces participants n'avaient eu aucune maladie infectieuse dans les 14 jours précédant l'essai, et aucune autre vaccination dans les 30 jours précédant l'essai. Les participants ont été répartis au hasard (3 contre 1) pour recevoir le vaccin ou le placebo, avec une stratification par groupe d'âge. Les enquêteurs, les participants et tout le personnel de l'étude ont été masqués pour l'affectation des groupes. Le vaccin a été administré (0,5 ml/dose) par voie intramusculaire selon un schéma "prime-boost" : un intervalle de 21 jours entre la première dose (rAd26) et la deuxième dose (rAd5). Le critère principal de l'essai était la proportion de participants présentant une COVID-19 confirmée par PCR à partir du 21e jour après avoir reçu la première dose. Toutes les analyses ont exclu les participants ayant enfreint le protocole. Le critère principal a été évalué chez les participants qui avaient reçu deux doses de vaccin ou de placebo. Les événements indésirables graves ont été évalués chez tous les participants qui avaient reçu au moins une dose au moment du verrouillage de la base de données, et les événements indésirables rares ont été évalués chez tous les participants qui avaient reçu deux doses et pour lesquels toutes les données disponibles ont été vérifiées dans le formulaire de déclaration de cas au moment du verrouillage de la base de données. L'essai est enregistré sur le site ClinicalTrials.gov (NCT04530396).
Résultats
Entre le 7 septembre et le 24 novembre 2020, 21 977 adultes ont été assignés au hasard au groupe vaccin (n=16 501) ou au groupe placebo (n=5476). 19 866 ont reçu deux doses de vaccin ou de placebo et ont été inclus dans l'analyse des résultats primaires. À partir du jour 21 après la première dose de vaccin (le jour de la deuxième dose), 16 (0,1%) des 14 964 participants du groupe vaccin et 62 (1,3%) des 4902 du groupe placebo avaient contracté la COVID-19. L'efficacité du vaccin était de 91,6% (95% CI 85-6-95-2). La plupart des effets indésirables signalés étaient de grade 1 (7 485 [94%] des 7 966 effets totaux). 45 (0,3%) des 16 427 participants du groupe vaccin et 23 (0,4%) des 5435 participants du groupe placebo ont eu des effets indésirables graves. Aucun n'a été considéré comme associé à la vaccination, avec confirmation par le comité indépendant de suivi des données. Quatre décès ont été signalés au cours de l'étude (3 [<0,1%] des 16 427 participants du groupe vaccin et 1 [<0,1%] des 5435 participants du groupe placebo), dont aucun n'a été considéré comme lié au vaccin.
Interprétation
Cette analyse intermédiaire de l'essai de phase 3 de Sputnik V a montré une efficacité de 91,6 % contre la COVID-19 et une bonne tolérance dans une large cohorte.
Financement
Département de la santé de la ville de Moscou, Fonds d'investissement direct russe, Sberbank et RUSAL.
Vaccin Novavax : NVX-CoV2373. Communiqué de presse (28 Janvier 2021)
NVX-CoV2373 est un candidat vaccin à base de protéine S (Spike) recombinante pleine longueur du SARS-CoV-2. Dans ce vaccin la protéine S est associée à un adjuvant breveté par Novavax, constitué de saponine - M™ (formation de nanoparticules) pour renforcer la réponse immunitaire afin de stimuler la production d'anticorps neutralisants. Le NVX-CoV2373 ne peut ni se répliquer, ni provoquer la COVID-19.
Plus de 37 000 participants ont participé à ce jour à quatre études cliniques différentes dans cinq pays. Le NVX-CoV2373 est actuellement évalué dans le cadre de deux essais cliniques de phase 3 : un essai au Royaume-Uni qui a terminé l'enrôlement en novembre et l'essai "PREVENT-19" aux États-Unis et au Mexique qui a commencé en décembre.
Novavax: résultats de la phase 3 au Royaume-Uni : 89,3 % d'efficacité (résultats non encore publiés).
Plus de 15 000 participants âgés de 18 à 84 ans, dont 27 % de plus de 65 ans, ont participé à l'étude. Ces participants étaient tous négatifs en PCR pour le SARS-CoV-2 au départ de l'étude. Le principal objectif de cet essai clinique était la première apparition d'une COVID-19 symptomatique (légère, modérée ou grave) confirmée par PCR dans un délai d'au moins 7 jours après la deuxième dose du vaccin.
La première analyse intermédiaire est basée sur 62 cas de COVID-19, dont 56 ont été observés dans le groupe placebo contre 6 dans le groupe NVX-CoV2373, ce qui donne une estimation ponctuelle de l'efficacité du vaccin de 89,3%. Sur les 62 cas, 61 étaient légers ou modérés et 1 était sévère (dans le groupe placebo).
L'analyse préliminaire indique que le variant britannique B.1.1.7, de plus en plus répandu, a été détecté dans plus de 50 % des 62 cas d'infection symptomatique confirmés par PCR (32 cas porteurs du variant britanniques, 24 porteurs du variant normal, et 6 cas d'un variant inconnu). Ainsi, l'efficacité pour le variant original était de 95,6 % et l'efficacité pour le variant britannique était de 85,6 %.
Les événements indésirables graves, sérieux et médicalement assistés se sont produits à des niveaux faibles et ont été équilibrés entre les groupes vaccinaux et placebo.
Par contre, Novavax a dévoilé que son vaccin NVX-CoV2373 ne serait efficace que sur moins de 50% des porteurs du variant B.1.135 détectée en Afrique du Sud et qui se propage dans le monde.
Keech et al. Phase 1–2 Trial of a SARS-CoV-2 Recombinant Spike Protein Nanoparticle Vaccine. New England Journal of Medicine 2020
Un premier essai randomisé de phase 1-2, contrôlé par placebo, pour évaluer la sécurité et l'immunogénicité du vaccin NVX-CoV2373 (en doses de 5μg et 25μg, avec ou sans adjuvant Matrix-M1, et avec des observateurs ignorant les affectations des groupes d'essai) a été conduit chez 131 adultes en bonne santé. La vaccination a consisté en deux injections intramusculaires, à 21 jours d'intervalle. Les premiers critères d'évaluation étaient la réactogénicité (test de sécurité), les valeurs de laboratoire (chimie du sérum et hématologie) selon les scores de toxicité de la Food and Drug Administration, et la production d'anticorps (IgG, test ELISA) contre la protéine S du virus. Les critères secondaires étaient les événements indésirables, la neutralisation du virus variant normal et les réponses des cellules T. Les résultats des tests d'IgG et de neutralisation ont été comparés à ceux trouvés dans des échantillons de sérum de convalescence de patients atteints de COVID-19, dont la plupart étaient symptomatiques. Mes analyses ont été réalisées au 35eme jour après la deuxième dose.
Résultats
Quatre-vingt trois participants ont été désignés pour recevoir le vaccin avec adjuvant, 25 ont reçu le vaccin sans adjuvant, et 23 participants ont été désignés pour recevoir le placebo. Aucun événement indésirable grave n'a été constaté. La réactogénicité était absente ou légère chez la majorité des participants, plus fréquente avec l'adjuvant, et de courte durée (moyenne, ≤2 jours). Un participant a eu une légère fièvre qui a duré 1 jour. Les événements indésirables étaient légers chez la plupart des participants ; il n'y a pas eu d'événements indésirables graves. L'ajout d'un adjuvant a entraîné une amélioration des réponses immunitaires, a permis d'économiser la dose d'antigène et a induit une réponse immunitaire de type Th1. La réponse Th1 est caractérisée par la production d' interféron-gamma, qui active les macrophages et induit les lymphocytes B à fabriquer des anticorps opsonisants (préparation pour la phagocytose) et de fixation du complément. Cette réponse conduit à une immunité à médiation cellulaire. Le schéma adjuvant à deux doses 5μg a induit des réponses moyennes des IgG anti-S (63 160 unités ELISA) et de neutralisation (3906 unités) qui ont dépassé les réponses moyennes obtenues avec le sérum de convalescence de patients COVID-19 pour la plupart symptomatiques (8344 et 983 unités, respectivement).
Conclusions
Au jour 35 de l'étude, le NVX-CoV2373 semblait être sans danger, et a provoqué des réponses immunitaires qui ont dépassé les résultats obtenus avec les sérums de convalescence. L'adjuvant Matrix-M1 a induit la production de lymphocytes T de phénotype Th1 (stimulation de la réponse immune, lymphocytes B et production d'anticorps IgM et IgG). (Financé par Epidemic Preparedness Innovations ; ClinicalTrials.gov, NCT04368988).
Vaccin Janssen et Johnson & Johnson (13 Janvier 2021)
Sadoff et al. Interim Results of a Phase 1–2a Trial of Ad26.COV2.S Covid-19 Vaccine. New England Journal of Medicine 2021
La compagnie pharmacetique Janssen (Johnson & Johnson) a développé un candidat vaccin appelé Ad26.COV2.S. Il s'agit d'un vaccin à vecteur adénoviral de sérotype 26 (Ad26), incapable de se répliquer et recombinant pour un ADN codant pour la protéine S (Spike) pleine longueur et stabilisée du SARS-CoV-2.
Méthode
Dans cet essai multicentrique (Belgique et Etats-Unis) de phase 1-2a, contrôlé par placebo, des adultes en bonne santé âgés de 18 à 55 ans (cohorte 1) et de 65 ans ou plus (cohorte 3) ont reçu le vaccin Ad26.COV2.S à une dose de 5×1010 particules virales (faible dose) ou de 1×1011 particules virales (forte dose) par millilitre ou ont reçu un placebo dans un schéma à une ou deux doses. Les principaux critères d'évaluation étaient la sécurité et la réactogénicité de chaque schéma posologique.
Résultats
Après l'administration de la première dose de vaccin à 805 participants des cohortes 1 et 3 et après la deuxième dose de la cohorte 1, les effets indésirables les plus fréquemment observés étaient la fatigue, les maux de tête, la myalgie et la douleur au point d'injection. L'événement indésirable systémique le plus fréquent était la fièvre. Les événements indésirables systémiques étaient moins fréquents dans la cohorte 3 que dans la cohorte 1 et chez les personnes ayant reçu la faible dose de vaccin que chez celles ayant reçu la forte dose. La réactogénicité était plus faible après la deuxième dose. Des anticorps neutralisants contre le virus de type sauvage ont été détectés chez 90 % ou plus de tous les participants au 29e jour après la première dose de vaccin (titre moyen géométrique 224 à 354) et chez 100 % des participants au 57e jour avec une nouvelle augmentation des titres (288 à 488), indépendamment de la dose de vaccin ou du groupe d'âge. Les titres des anticorps neutralisants sont restés stables au moins jusqu'au jour 71. Une deuxième dose a permis d'augmenter le titre d'un facteur de 2,6 à 2,9 (827 à 1266). La réponse des anticorps se liant à la protéine S était similaire à celle des anticorps neutralisants. Le 14e jour, des réponses des cellules T CD4+ ont été détectées dans 76 à 83 % des participants de la cohorte 1 et de 60 à 67 % de ceux de la cohorte 3, avec une nette asymétrie vers les cellules T auxiliaires de type 1. Les réponses des cellules T CD8+ ont été globalement robustes mais plus faibles dans la cohorte 3.
Conclusions
Les profils de sécurité et d'immunogénicité de l'Ad26.COV2.S soutiennent la poursuite du développement de ce candidat vaccin. (Financé par Johnson & Johnson et le Biomedical Advanced Research and Development Authority of the Department of Health and Services à la personne; COV1001 numéro ClinicalTrials.gov, NCT04436276). L’essai de phase 3 a démarré le 1er février et doit durer deux ans : 30 000 personnes y participent dans le monde, dont 1175 en France. Ce vaccin ne devrait pas être le seul à être éprouvé par les volontaires recrutés par l’Inserm par le biais de sa plateforme, Covireivac. A suivre...
Vaccin Pfizer-BioNTech: des cas (très rares) d'anaphylaxie signalés (fin décembre 2020)
Selon le CDC (Centre for Disease Control), entre le 11 Décembre 2020, date à laquelle le premier vaccin anti-COVID a été autorisé aux Etats-Unis, et le 23 Décembre 2020, 1 893 860 personnes ont été vaccinées. Vingt et un cas d'anaphylaxie ont été signalés (un taux de 11,1 par million de doses administrées, ou 1 cas pour 100 000), dont 17 chez des personnes ayant présenté des antécédents documentés d'allergies ou de réactions allergiques, dont sept avaient des antécédents d'anaphylaxie. Aucun cas mortel n'a été observé. L'intervalle médian entre la réception du vaccin et l'apparition des symptômes était de 13 minutes (intervalle = 2-150 minutes). Parmi les 20 personnes pour lesquelles des informations de suivi étaient disponibles, toutes s'étaient rétablies ou avaient été renvoyées chez elles. Toute réaction suspecte d'anaphylaxie doit être traitée immédiatement par une injection intramusculaire d'épinéphrine (adrénaline).
Ces réactions anaphylactiques pourraient être dues au polyéthylène glycol (PEG) utilisé pour stabiliser les nanoparticules lipidiques qui protègent l'ARNm des vaccins Pfizer et Moderna (de Vrieze. Pfizer’s vaccine raises allergy concerns. Polymer in mRNA’s “packaging” may cause rare anaphylactic reactions. Science 2021; Castells et al. Maintaining Safety with SARS-CoV-2 Vaccines. New England Journal of Medicine 2021). Les PEG sont utilisés depuis des décennies comme épaississants dans les produits courants comme le dentifrice, le shampoing, certains solvants et les laxatifs. Un nombre croissant de produits biopharmaceutiques en contiennent également. On a longtemps pensé que ces composés étaient biologiquement inerte, mais les preuves s'accumulent qu'ils ne le sont pas. Cependant, la quantité de PEG présente dans le vaccin est beaucoup plus faible que celle relative aux produits cosmétiques ou pharmaceutiques. Quoiqu'il en soit, le CDC et la FDA vont renforcer leur surveillance sur les cas possibles d'anaphylaxie chez les personnes ayant reçu le vaccin COVID-19.
Le document du CDC du 6 Janvier 2021 est téléchargeable ici: ![]() Cdc anaphylacsis after pfizer vaccine in usa january 6 2021 copie (240.74 Ko).
Cdc anaphylacsis after pfizer vaccine in usa january 6 2021 copie (240.74 Ko).
A suivre...
Vaccin Pfeizer-BioNTech. Publication dans le New England Journal of Medicine 2021 (31 Décembre 2020)
Polack et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. NEW England Journal of Medicine 2020.
Le vaccin à ARNm (BNT162b2) codant pour la protéine S pleine-longueur du SARS-CoV-2 a déjà été présenté sur ce site, voir page "La course aux vaccins-Vaccins à ARN).
Méthodes
Dans cet essai d'efficacité multinational contrôlé par placebo et en aveugle, des personnes âgées de 16 ans ou plus ont été réparties dans un rapport de 1:1 pour recevoir deux doses, à 21 jours d'intervalle, soit de placebo, soit du candidat vaccin BNT162b2 (30 μg par dose). Les principaux critères d'évaluation étaient l'efficacité du vaccin contre la Covid-19 confirmé en laboratoire (RT-PCR) et la sécurité. Les participants potentiels ayant eu des antécédents de réaction allergique à l'un des composants du vaccin ont été exclus.
Résultats
Un total de 43 548 participants ont été randomisés, dont 43 448 ont reçu des injections : 21 720 avec le BNT162b2 et 21 728 avec le placebo. Il y a eu 8 cas de Covid-19 avec apparition au moins 7 jours après la deuxième dose parmi les participants assignés à recevoir le BNT162b2 et 162 cas parmi ceux assignés au placebo ; le BNT162b2 était efficace à 95% pour prévenir la Covid-19. Une efficacité similaire du vaccin (généralement de 90 à 100 %) a été observée dans les sous-groupes définis par l'âge, le sexe, la race, l'origine ethnique, l'indice de masse corporelle de base et la présence d'affections coexistantes. Parmi les 10 cas de Covid-19 grave apparus après la première dose, 9 sont survenus chez des receveurs de placebo et 1 chez un receveur de BNT162b2. Le profil de sécurité du BNT162b2 était caractérisé par une douleur de courte durée, légère à modérée, au point d'injection, de la fatigue et des maux de tête. L'incidence des effets indésirables graves était faible et similaire dans les groupes vaccin et placebo.
Conclusion
Un schéma de deux doses de BNT162b2 a conféré une protection de 95 % contre Covid-19 chez les personnes âgées de 16 ans ou plus. La sécurité sur une période médiane de 2 mois était similaire à celle des autres vaccins viraux. (Financé par BioNTech et Pfizer ; numéro de ClinicalTrials.gov, NCT04368728).
Vaccin Moderna. Publication dans le New England Journal of Medicine 2021 (30 Décembre 2020)
Baden et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. New England Journal of Medicine 2021.
Le vaccin à ARNm (mARN-1273, développé par Moderna) a déjà été présenté sur ce site, voir page "La course aux vaccins-Vaccins à ARN).
Cet article présente les résultats de l'essai randomisé de phase 3, en aveugle et contrôlé par placebo, mené dans 99 centres à travers les États-Unis. Les personnes présentant un risque élevé d'infection par le SARS-CoV-2 ou de ses complications. Le principal critère d'évaluation était la prévention de la Covid-19, avec une apparition au moins 14 jours après la deuxième injection chez les participants qui n'avaient pas été infectés auparavant par le SARS-CoV-2. Les participants potentiels ayant eu des antécédents de réaction allergique à l'un des composants du vaccin ont été exclus.
Résultats
L'essai a recruté 30 420 volontaires répartis au hasard dans un rapport de 1:1 pour recevoir soit le vaccin, deux injections intramusculaires d'ARNm-1273 (100 μg), soit le placebo (15 210 participants dans chaque groupe). Plus de 96 % des participants ont reçu les deux injections, et 2,2 % présentaient des signes (sérologiques, virologiques ou les deux) d'infection par le SARS-CoV-2 au départ de l'essai. La maladie Covid-19 symptomatique a été confirmée chez 185 participants du groupe placebo et chez 11 participants du groupe vaccin ARNm-1273; l'efficacité du vaccin était de 94,1% (P<0,001). L'efficacité était similaire dans les principales analyses secondaires, y compris l'évaluation 14 jours après la première dose, les analyses incluant les participants qui avaient des preuves d'une infection par le SARS-CoV-2 au départ, et les analyses chez les participants âgés de 65 ans ou plus. Des cas graves de Covid-19 sont survenus chez 30 participants, avec un décès ; les 30 participants faisaient tous partie du groupe placebo. Une réaction modérée et transitoire après la vaccination s'est produite plus fréquemment dans le groupe vaccin. Les effets indésirables graves étaient rares, et l'incidence était similaire dans les deux groupes.
Conclusions
Le vaccin ARNm-1273 a montré une efficacité de 94,1 % dans la prévention de la Covid-19, y compris des graves graves. Hormis les réactions locales et systémiques transitoires, aucun problème de sécurité n'a été identifié. (Cette étude a été financé par la Biomedical Advanced Research and Development Authority et le National Institute of Allergy and Infectious Diseases ; COVE ClinicalTrials.gov, NCT04470427).
Vaccin Pfeizer-BioNTech: document fourni à la FDA (10 décembre 2020)
Ce document (en Anglais, 92 pages) fourni par le groupe Pfeizer à la FDA le 30 Novembre 2020 est téléchargeable ici : ![]() Vaccin pfeizer fda vrbpac 12 10 20 meeting briefing document sponsor (1.65 Mo).
Vaccin pfeizer fda vrbpac 12 10 20 meeting briefing document sponsor (1.65 Mo).
Ci-dessous la figure 13 du document (page 58).
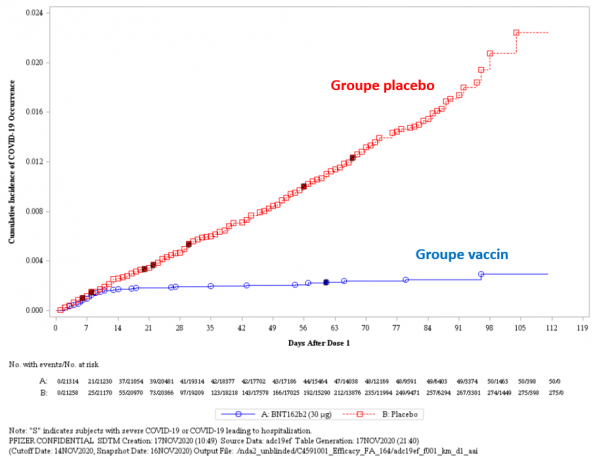
Courbes d'incidence de la COVID-19 après la première dose du vaccin dans les groupes placebo et vaccin.
La vaccination a eu lieu au jour 0 et les cas d'apparition de la COVID-19 (test RT-PCR positif) ont été enregistrés en fonction du temps dans les deux groupes de volontaires: environ 22000 dans chaque groupe. On voit que jusqu'au jour 14 environ les deux courbes sont parallèles. A partir du jour 14, elles se séparent nettement. L'efficacité du vaccin se manifeste 14 jours après la vaccination. Au total 10 cas de COVID-19 graves (ayant nécessité une hospitalisation) sont apparu après le jour 0, soient 9 dans le groupe placebo (carrés rouges) et un dans le groupe vaccin (carré bleu foncé). Parmi tous les volontaires, 50 cas de COVID-19 sont apparus dans le groupe vaccin et 275 dans le groupe placebo. La protection apportée par le vaccin semble donc efficace dans les atteintes graves et non graves.
Que s'est-il passé avec le vaccin Australien ? (11 Décembre 2020)
Vendredi 11 Décembre dernier, le premier ministre Australien Scott Morrison et son ministre de la santé Greg Hunt annonçaient que les essais cliniques de phase trois du vaccin anti-COVID-19 développé par l'Université du Queensland à Brisbane étaient définitivement stoppés. De fait, la décision du gouvernement fédéral Australien d'acheter plus de 50 millions de doses du vaccin potentiel contre le SARS-CoV-2 a été brutalement annulée. Le motif officiellement invoqué était que des résultats faussement positifs à certains tests de dépistage du VIH (virus du SIDA) avaient été enregistrés chez les participants de l'essai de phase 2 en cours.
Explications
Ce vaccin a été développé par l'équipe de Paul Young (Université du Queensland, Brisbane), co-directeur du projet de vaccin, sur la base d'une nouvelle technologie mise au point dans son laboratoire : "la pince moléculaire" ("molecular clamp", en Anglais).
A l'inverse des vaccins traditionnels à base de virus inactivé ou rendu inoffensif par diverses manipulations, il s'agit d'un vaccin sous-unitaire, c'est à dire ne contenant qu'un petit fragment du virus SARS-CoV-2, en l'occurrence la protéine de pointe (Spike), appelée aussi protéine S. Bien évidemment, il est totalement impossible que l'injection de cette protéine virale à un humain puisse provoquer la maladie.
Dans le SARS-CoV-2, les protéines S, également appelées protéines de fusion, se trouvent régulièrement réparties à la surface de la membrane du virus et c'est par elles que s'établit d'une part le contact avec les cellules humaines (ou animales) dans lesquelles le virus va pénétrer, et d'autre part le processus de fusion entre la membrane virale et la membrane de la cellule hôte afin que le génome viral puisse être injecté dans la cellule.
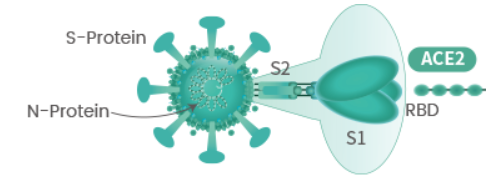
Schéma d'un trimère de la protéine S du SARS-CoV-2
Dans la membrane du virus les protéines S s'assemblent en trimères (trois protéines "collées" par des interactions non covalentes). Ce sont ces trimères qui assurent l'interaction entre la membrane virale (à gauche), assurée par la partie S2 et la membrane cellulaire assurée par la liaison de la partie S1 avec le récepteur ACE2 lui-même ancré dans la membrane cellulaire (à droite non figurée). (Schéma selon SinoBiological).
Si le système immunitaire reconnaît cette protéine S, il va produire des anticorps neutralisants qui vont bloquer le virus et le détruire. C'est évidemment le but d'un vaccin.
Ce type de vaccin est cependant confronté à un défi majeur : les protéines de fusion telles que la protéine S du SARS-CoV-2 changent de forme lorsqu'elles se lient aux cellules humaines et fusionnent avec la paroi cellulaire. Il est important de comprendre qu'à la surface du virus toutes ces protéines de fusion se trouvent sous une forme métastable dite de "pré-fusion". Au moment où s'établit le contact entre le virus et son récepteur sur la membrane cellulaire, les protéines de fusion subissent un changement conformationnel majeur vers une forme dite de "post-fusion". Le repliement dans cette forme "post-fusion" très stable déclenche le processus de fusion entre la membrane du virus et la membrane de la cellule hôte. Le changement conformationnel impliqué dans cette transition modifie de façon spectaculaire la topographie de surface de la protéine S et donc ses épitopes antigéniques, c'est à dire les motifs moléculaires qu'elle présente aux cellules du système immunitaire. Autrement dit, les épitopes antigéniques de la protéine S dans sa conformation "pré-fusion" sont différents des épitopes antigéniques présentés dans sa conformation "post-fusion". Par exemple, la configuration "pré-fusion" expose des parties de la protéine S cachées dans sa forme "post-fusion" et peut générer une réponse anticorps plus robuste.
Protéine S en pré- et post-fusion
Images obtenues en cryo-microscopie électronique. A gauche la partie de la protéine S du SARS-CoV-2 qui participe au processus de fusion (en couleur) entre les deux membranes virale et cellulaire dans sa conformation "pré-fusion". A droite la même protéine dans sa conformation "post-fusion" (Selon Fan et al. Nature Comm 2020).
Or, pour provoquer une réponse immunitaire forte qui peut empêcher l'infection, les anticorps induits doivent reconnaître les protéines virales dans leur conformation active, "pré-fusion", celle qu'elles présentent naturellement à la surface du virion. De fait toutes les études montrent que les anticorps neutralisants générés par le système immunitaire contre les virus tels que le SARS-CoV-2 sont naturellement dirigés contre les protéines S dans leur forme naturelle, c'est à dire la conformation "pré-fusion".
Si on veut produire un vaccin efficace contre le virus il faut donc être capable de deux choses:
- produire la protéine S de fusion pour l'injecter aux personnes à vacciner
- faire en sorte que cette protéine de fusion se trouve dans la configuration "pré-fusion", sa configuration naturelle.
Le problème est que dans leur forme purifiée ou isolée (c'est à dire hors du virus) les protéines de fusion sont naturellement instables et adoptent la conformation de "post-fusion", thermodynamiquement plus stable. Et c'est là qu'entre en jeu le concept de "pince moléculaire" mis au point par l'équipe de Paul Young (Wijesundara et al., Frontiers in Immunology 2020).
La pince moléculaire (molecular clamp en Anglais)
Produire la protéine S est facile à partir du moment où on connait la séquence du génome viral. En revanche, ce qui est plus compliqué est de la produire dans la configuration "pré-fusion". La pince moléculaire a été conçue pour résoudre ce problème. Elle utilise des peptides (petits fragments de protéine) fixés sur la base (partie S2) de la protéine S pour la maintenir dans sa forme "pré-fusion". La présence de tels peptides dans la séquence de la protéine S permettrait alors au système immunitaire de générer une réponse antivirale potentiellement plus forte.
C'est la pari qu'a fait l'équipe de Paul Young.
Après une longue recherche au cours de laquelle plusieurs peptides différents ont été testés, les meilleurs étaient ceux présents dans la protéine GP41 du virus du SIDA, le VIH. La protéine GP41 permet la fusion entre la membrane du VIH et la membrane de sa cellule hôte.
Les chercheurs ont donc construit une protéine S recombinante (SARS-CoV-2-S-Clamp), c'est à dire produite par génie génétique (à partir de la séquence de son ARN), dans laquelle les peptides de fusion de la protéine GP41 ont été introduits comme indiqué dans le schéma ci-dessous.
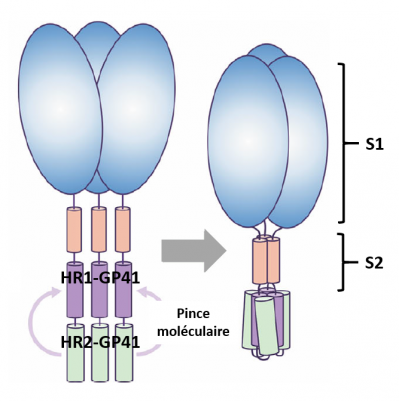
Schéma de la protéine SARS-CoV-2-SClamp
A gauche, schéma d'un trimère de la protéine S isolée purifiée produite par génie génétique. Elle se trouve dans une conformation "post-fusion". A droite, le trimère de la même protéine hybride, couplée à deux peptides HR1 et HR2 de fusion de la protéine du VIH GP41. Ces deux peptides confèrent à la protéine SClamp une conformation pré-fusion plus immunogène (selon Wijesundara et al., Frontiers in Immunol 2020).
Le vaccin Australien
Cette protéine S-Clamp a été utilisée pour produire le vaccin Australien. Testé sur divers modèles animaux (souris, hamster et rat) ce vaccin a donné d'excellents résultats en termes de sécurité, de production d'anticorps neutralisants et de réponse des lymphocytes cytotoxiques CD4+ and CD8+.
Concevoir un vaccin qui contenait un fragment du VIH sans déclencher la production d'anticorps anti-VIH allait s'avérer difficile. Pour ce faire, l'équipe Australienne a soigneusement supprimé les sites (épitopes) connus de la protéine GP41 du VIH auxquels les anticorps se lient. L'espoir était que cela suffirait pour que le vaccin ne génère pas d'anticorps anti-VIH, mais uniquement des anticorps anti-protéine S du SARS-CoV-2.
Lors des premiers tests sur les modèles animaux, tout semblait fonctionner. Mais le problème allait apparaître chez les humains. En effet, les tests standards de détection du VIH ne fonctionnent pas sur les animaux, ce qui signifie que les chercheurs ne pouvaient pas vérifier si leur vaccin produisait des anticorps anti-VIH avant le début des essais cliniques chez l'homme.
Lorsque les 216 participants à l'essai de phase 1 se sont inscrits pour participer à l'étude, ils ont été informés par les chercheurs que des faux positifs pour le VIH pouvaient apparaître et des examens sanguins ont été programmés pour détecter et surveiller l'apparition éventuelle de ces anticorps anti-VIH. Chaque participant qui avait reçu le vaccin a été testé faiblement positif sur certains tests VIH standards conçus pour rechercher spécifiquement des anticorps dirigés contre la protéine d'enveloppe du VIH, la glycoprotéine GP41.
On ne sait pas encore exactement pourquoi ces anticorps sont apparus, étant donné que l'équipe avait soigneusement retiré les sites de liaison d'anticorps probables de la protéine du VIH.
- L'apparition de ces anticorps n'est en aucun cas un risque pour la santé des participants.
- Ils n'ont pas contracté le VIH.
- Stimulé par le vaccin, leur système immunitaire a généré des anticorps contre le VIH.
Il est tout simplement regrettable que ces anticorps soient exactement ceux que recherchent les tests courants de détection du VIH.
L'équipe espérait que les niveaux de ces anticorps chuteraient rapidement, mais cela n'a pas été le cas. Ils ont exploré d'autres options avec des spécialistes du VIH mais sans succès. Et en fin de compte, il a été décidé d'arrêter le développement de ce vaccin parce qu'il allait interférer de façon inacceptable avec le test de dépistage du VIH pour la population Australienne.
Mis à part la réponse immunitaire problématique sur la protéine GP41 du VIH, le vaccin administré aux 216 participants en juillet 2020, avait montré une réponse robuste contre le SARS-CoV-2 et un profil d'innocuité solide. L'essai d'innocuité de phase I se poursuivra, pour voir combien de temps les anticorps dirigés contre la protéine GP41 du VIH persistent, même si les indications sont que les niveaux diminuent déjà. L'université prévoit de soumettre les données complètes pour publication à comité de lecture.
A l'issu de la découverte des résultats et de l'annonce du gouvernement de suspendre définitivement sa commande de 50 millions de doses du vaccin, Paul Young déclarait: « Je pense qu'il y a un seul mot qui résume tout : dévasté! Les dernières 24 heures ont été particulièrement difficiles pour l'équipe. C'est une période difficile, mais c'est de la science. Cela ne fonctionne pas toujours ».
Mon analyse de ces résultats
- Ce vaccin était constitué par la protéine S du SARS-CoV-2 modifiée par la présence de deux peptides dérivés de la protéine GP41 de fusion du VIH afin de lui conférer une conformation proche (sinon identique) de celle de la pré-fusion, théoriquement plus immunogène. Cette stratégie laissait présager que des anticorps anti-GP41 seraient exprimés chez les personnes vaccinées. Les chercheurs avaient anticipé cette éventualité en essayant de supprimer certains épitopes sur ces deux peptides issus de la protéine GP41 du VIH. Ils n'ont manifestement pas réussi.
- Les personnes vaccinées n'ont pas contracté le SIDA. Simplement, elles ont été immunisées contre le VIH de façon "accidentelle" en plus de leur immunisation contre le SARS-CoV-2.
- Il est regrettable que la détection de ces anticorps anti-GP41 fassent partie de l'arsenal des tests standards de détection du VIH.
- Dans les autres stratégies vaccinales en particulier celles de Pfizer-BioNtech et Moderna, c'est la protéine S du SARS-CoV-2 "naturelle" (non modifiée) qui est utilisée pour activer le système immunitaire. La différence est qu'avec ces deux vaccins la protéine est produite directement dans les cellules via son ARN messager. Quelle conformation adopte-t-elle dans ces conditions: pré-fusion ou post-fusion ? Théoriquement, étant produite seule, en absence d'enveloppe virale, elle devrait adopter la conformation "post-fusion" (thermodynamiquement la plus stable). Le fait que ces vaccins s'avèrent efficaces à plus de 90% suggère soit que la conformation de cette protéine n'a pas un effet important sur la réponse immunitaire quelle induit chez l'homme, soit que les vaccins à ARN produisent une protéine S dans sa conformation pré-fusion dans les cellules humaines.
- Il est à noter que le vaccin développé par Sanofi et GSK est également basé sur la protéine S du SARS-CoV-2 recombinante, complétée avec un adjuvant dont je ne connais pas la nature. On sait que les premiers résultats de phase 1-2 obtenus avec ce vaccin sont en deçà de ce qui était attendu. Serait-ce la conséquence du fait que cette protéine adopte une configuration "post-fusion" dans le vaccin Sanofi-GSK ?
A suivre…
Vaccin AstraZeneca, AZD1222. Nouvelle publication dans le Lancet du 8 Décembre 2020
Voysey et al. Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: an interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. Lancet 8 Décembre 2020
La sécurité et l'efficacité du vaccin ChAdOx1 nCoV-19 ont été évaluées dans une analyse intermédiaire commune de quatre essais.
Méthodes
Cette analyse comprend des données provenant de quatre essais contrôlés, randomisés et en aveugle en cours au Royaume-Uni, au Brésil et en Afrique du Sud. Les participants âgés de 18 ans et plus ont été assignés de manière aléatoire (1:1) au vaccin ChAdOx1 nCoV-19 ou au contrôle (vaccin antiméningococcique conjugué des groupes A, C, W et Y ou solution saline). Les participants du groupe ChAdOx1nCoV-19 ont reçu deux doses contenant 5 × 10¹⁰ particules virales (dose standard ; cohorte SD/SD) ; un sous-ensemble dans l'essai britannique a reçu une demi-dose comme première dose (faible dose) et une dose standard comme deuxième dose (cohorte LD/SD). L'analyse primaire de l'efficacité a inclus la COVID-19 symptomatique chez des participants séronégatifs avec un frottis positif au test RT-PCR plus de 14 jours après une deuxième dose de vaccin. Les participants ont été analysés en fonction du traitement reçu, avec une date limite de collecte des données fixée au 4 novembre 2020. L'efficacité du vaccin a été calculée selon un modèle de régression de Poisson robuste ajusté en fonction de l'âge. Les études sont enregistrées sur ISRCTN89951424 et ClinicalTrials.gov, NCT04324606, NCT04400838, et NCT04444674.
Résultats
Entre le 23 avril et le 4 novembre 2020, 23 848 participants ont été inscrits et 11 636 participants (7548 au Royaume-Uni, 4088 au Brésil) ont été inclus dans l'analyse primaire d'efficacité intermédiaire. Chez les participants ayant reçu deux doses standard, l'efficacité du vaccin était de 62,1% et chez les participants ayant reçu une faible dose suivie d'une dose standard, l'efficacité était de 90,0%. L'efficacité globale du vaccin dans les deux groupes était de 70,4%. À partir de 21 jours après la première dose, 10 cas ont été hospitalisés pour COVID-19, tous dans le groupe témoin ; deux ont été classés comme COVID-19 grave, dont un décès. En termes de sécurité, 175 événements indésirables graves sont survenus chez 168 participants, 84 dans le groupe ChAdOx1 nCoV-19 et 91 dans le groupe témoin. Trois événements ont été classés comme pouvant être liés à un vaccin : un dans le groupe ChAdOx1 nCoV-19, un dans le groupe de contrôle et un chez un participant qui reste masqué à l'attribution du groupe.
Interprétation
Le ChAdOx1 nCoV-19 a un profil de sécurité acceptable et s'est révélé efficace contre symptomatique COVID-19 dans cette analyse intermédiaire des essais cliniques en cours.
Vaccin AstraZeneca, AZD1222. Communiqué de presse du 19 Oct 2020 (AstraZeneca)
J’ai déjà parlé de ce vaccin développé dans le cadre d’une collaboration entre l’Université d’Oxford (Royaume Unis) et AstraZeneca (Cambridge, Royaume Unis). Voir plus bas : ChAdOx1 nCoV-19 ou AZD1222. 20 Juillet 2020.
Il s’agit d’un vaccin à base de vecteur viral dans lequel un adénovirus recombinant de Chimpanzé exprime la protéine S du SARS-CoV-2.
Pourquoi un adénovirus de Chimpanzé ?
Parce que nous sommes tous susceptibles d’avoir été infectés par divers types d’adénovirus humains et que nous pourrions donc posséder des anticorps contre ces adénovirus. Ces anticorps pourraient interférer avec le vaccin et inhiber son action. Par contre, nous ne possédons pas d’anticorps dirigés contre des adénovirus d’autres espèces animales.
Le vaccin ChAdOx1 est un vecteur de vaccin adénoviral de Chimpanzé. Il s'agit d'un adénovirus inoffensif et affaibli qui provoque généralement le rhume chez le Chimpanzé. Le ChAdOx1 a été choisi comme la technologie la plus appropriée pour un vaccin contre le SARS-CoV-2, car il a été démontré qu'il génère une forte réponse immunitaire. Il a été génétiquement modifié de sorte qu'il est impossible qu'il se développe chez l'homme. Il est donc plus sûr de le donner aux enfants, aux personnes âgées et à toute personne souffrant d'une maladie préexistante comme le diabète. Les vecteurs adénoviraux du chimpanzé sont un type de vaccin très bien étudié, ayant été utilisé en toute sécurité chez des milliers de sujets contre un certain nombre d'agents pathogènes, dont la grippe, le Zika et le syndrome respiratoire du Moyen-Orient (MERS) (Folegati et al. Lancet 2020 ; Tapia et al. Lancet Infect Dis 2020; Coughlan et al. EBioMedicine 2018; Wilkie et al. Vaccine, 2020).
Les résultats provisoires de l'essai de phase II/III COV002 en cours sur l'AZD1222 au Royaume-Uni, viennent d’être publiés dans le Lancet du 19 Novembre 2020.
Ramasamy et al. Safety and immunogenicity of ChAdOx1 nCoV-19 vaccine administered in a prime-boost regimen in young and old adults (COV002): a single-blind, randomised, controlled, phase 2/3 trial. Lancet 2020.
COV002 est un essai de phase II/III contrôlé, randomisé, multicentrique et en aveugle qui évalue la sécurité, l'efficacité et l'immunogénicité de l'AZD1222 par rapport à un placebo pour la prévention de la COVID-19, chez 12 390 participants au Royaume-Uni. Les participants à l'essai à ce jour sont âgés de 18 ans ou plus, sont en bonne santé ou ont des maladies chroniques médicalement stables et sont susceptibles d'être exposés au virus du SARS-CoV-2. Ils sont répartis aléatoirement pour recevoir une ou deux doses intramusculaires du vaccin de comparaison AZD1222 ou MenACWY (vaccin contre un méningocoque). La randomisation est stratifiée par âge, ≥18 à 55 ans, 56-69 ans et ≥70 ans, dans un rapport de 1:1, 3:1 ou 5:1, respectivement. Dans l'essai de phase II, les participants ont reçu soit une ou deux administrations d’une dose standard (3,5-6,55 x1010 particules virales), soit une dose faible (2,2 x1010 particules virales).
Les résultats montrent que les réactions locales et systémiques sont moins importantes chez les adultes âgés (≥56-69 ans et ≥70 ans) que chez les adultes plus jeunes (≥18-55 ans) et génèrent des réponses immunitaires robustes similaires contre le SARS-CoV-2 dans toutes les tranches d'âge adultes. Dans le groupe AZD1222 ces réactions sont comparables aux données provisoires précédemment rapportées avec le vaccin de l'essai de phase I/II et d'autres vaccins à vecteur adénoviral (Folegati et al. Lancet 2020 ; Tapia et al. Lancet Infect Dis 2020; Coughlan et al. EBioMedicine 2018; Wilkie et al. Vaccine, 2020). Les réactions ont été moins nombreuses après une deuxième dose d'AZD1222 et avec la dose inférieure. La fréquence des réactions diminuait également avec l'âge.
Les réponses des anticorps à la protéine de pointe du virus du SRAS-CoV-2 ont été détectées au 28e jour chez 100 % des participants, indépendamment de l'âge ou de la dose de vaccin, et ont continué à augmenter après une deuxième dose d'AZD1222. Une activité neutralisante a été observée chez 100 % des participants au 14e jour après avoir reçu une deuxième dose standard, quel que soit leur âge. Des réponses des cellules T cytotoxiques ont été induites, atteignant un pic au 14e jour après la première dose et se maintenant deux mois après l'injection, quels que soient l'âge (notamment chez les sujets âgés, > 70 ans) et le dosage.
Des essais cliniques de phase 3 de l'AZD1222 sont en cours dans le monde entier au Royaume-Uni, aux États-Unis, au Brésil, en Afrique du Sud, au Japon, en Russie et au Kenya, avec jusqu'à 60 000 participants dans une large tranche d'âge et dans divers groupes raciaux, ethniques et géographiques. Les premiers résultats des essais en phase finale sont attendus avant la fin de l'année, en fonction du taux d'infection par le SARS-CoV-2 au sein des communautés participant aux essais.
AstraZeneca s’est engagé à respecter son engagement à soutenir un accès large et équitable au vaccin, sans profit, pendant la pandémie, si les essais cliniques s'avèrent fructueux et sont approuvés par les autorités de réglementation. La capacité d'approvisionnement mondiale a été assurée pour trois milliards de doses, produites sur douze chaînes d'approvisionnement régionales et impliquant plus de vingt organisations de fabrication sous contrat, y compris les sites de fabrication d'AstraZeneca. Des progrès rapides sont réalisés dans tous les aspects de la fabrication du vaccin, de la substance médicamenteuse au conditionnement en passant par la formulation et le remplissage. Il est important d'aligner la livraison du produit pharmaceutique final sur l'approbation réglementaire pour garantir une durée de conservation restante maximale.
Vaccin Pfizer-BioNtech : communiqué de presse du 18/11/2020 (sur Businesswire)
L'essai de Pfizer et BioNTech qui a inclus près de 44 000 volontaires se poursuit. Dans cet essai, la moitié a reçu le vaccin et l'autre moitié a reçu une injection placebo d'eau salée. Les chercheurs attendent de voir combien de personnes de chaque groupe va développer la COVID-19.
Pfizer et BioNTech ont déclaré que sur 170 cas de Covid-19 qui sont apparus depuis le début de l’essai, 162 faisaient partie du groupe placebo et 8 du groupe vaccin. Le taux d'efficacité passe à 95%. Sur dix cas de Covid-19 grave, neuf avaient reçu un placebo. Dans le premier communiqué (9/11/2020) les chiffres étaient de 94 avec 5 cas de COVID dans le groupe vaccin.
Pfizer et BioNTech ont déclaré que l'efficacité du vaccin était constante quel que soit l'âge, la race et l'origine ethnique. L'effet indésirable grave le plus fréquent était la fatigue, 3,7 % des volontaires ayant déclaré être fatigués après avoir pris la deuxième dose. Deux pour cent des volontaires ont déclaré avoir eu mal à la tête après la deuxième dose. Les adultes plus âgés ont signalé des effets secondaires moins nombreux et plus légers.
En plus des résultats de son essai clinique, Pfizer a déclaré qu'elle était prête à soumettre à la FDA deux mois de données de sécurité que l'agence avait recommandées, ainsi que des dossiers de fabrication détaillés montrant que l'entreprise peut produire de façon constante des lots de haute qualité de son produit. La FDA examinera les données et demandera à un groupe externe d'experts en vaccins de se prononcer sur la demande, un processus qui pourrait prendre des semaines.
Si le vaccin est autorisé, l'attention se portera immédiatement sur la manière dont il sera distribué. Le vaccin doit être stocké à -80°C, c'est-à-dire plus froid que tout autre vaccin en cours de développement. Pfizer expédiera le vaccin dans des boîtes spéciales de 1 000 à 5 000 doses qui seront remplies de glace carbonique et équipées de capteurs GPS. Le vaccin de Pfizer peut être stocké dans des congélateurs classiques pendant cinq jours au maximum, ou dans les glacières spéciales pendant 15 jours au maximum, à condition que la glace sèche soit renouvelée et que les boîtes ne soient pas ouvertes plus de deux fois par jour.
Le vaccin de Moderna doit également être stocké dans un congélateur à long terme, mais à moins 4 degrés Fahrenheit. La société a déclaré lundi que son vaccin pouvait être stocké à des températures standard de 36 à 46 degrés Fahrenheit au réfrigérateur pendant 30 jours, et non sept comme on le pensait auparavant, ce qui pourrait le rendre plus facile à stocker que le vaccin Pfizer.
Pourquoi ces basses températures ?
Les vaccins Moderna et Pfizer-BioNTech fournissent aux cellules de l'organisme un ARNm qui code pour la fabrication de la protéine de pointe (Spike) du SARS-CoV-2, le virus qui provoque la COVID-19 (Voir ici sur ce site). La protéine migre ensuite à la surface de la cellule et déclenche une réponse immunitaire. L'ARNm est relativement fragile par rapport aux protéines ou aux fragments de protéines qui composent souvent les vaccins classiques, et il se dégrade facilement à température ambiante. De plus, les enzymes appelées ribonucléases qui dégradent les ARNm "sont présentes partout, même dans l'environnement contrôlé d'un laboratoire". Les chercheurs en laboratoire et les entreprises protègent l'ARNm pendant la production et le stockage en l'insérant dans un support, une sorte de sac, substance semblable à du gras appelée nanoparticule lipidique (également appelés liposomes). Ces particules lipidiques protègent l'ARNm des enzymes présentes dans le sang une fois qu'il a été injecté. Mais la nanoparticule est délibérément conçue pour se dégrader lentement, afin qu'elle ne s'accumule pas dans le foie et ne cause pas de dommages.
mRNA1273. 16 Novembre 2020.
Vaccin à ARN déjà présenté sur ce site (voir ci-dessous) le 14 Juillet 2020
mRNA1273 est un vaccin à ARN développé par Moderna, une compagnie américaine basée à Cambridge MA, USA, en collaboration avec l'Institut National des Allergies et des Maladies Infectieuses (NIAID). Ce candidat vaccin a déjà été présenté sur ce site (voir ci-dessous) le 14 Juillet 2020. Moderna a été l'un des premiers développeurs de vaccin à ARN et à annoncer qu'il travaillait sur un vaccin COVID-19 et à passer aux essais cliniques chez l'homme. Son vaccin est constitué d'un ARN codant pour une forme modifiée de la protéine S (Spike) du SARS-CoV-2, la principale cible du système immunitaire contre ce coronavirus, encapsulé dans des nanoparticules lipidiques qui en assurent la stabilité et la délivrance dans l'organisme. Un essai de phase 3 a débuté le 27 juillet et a recruté environ 30 000 personnes. Cet essai se poursuit. Mais une analyse menée le 15 novembre par un Comité Indépendant de Sécurité des Données a révélé que 95 participants à l'essai avaient développé la COVID-19. Parmi eux, 90 faisaient partie du groupe ayant reçu une injection de placebo et 5 avaient reçu le vaccin, ce qui équivaut à une efficacité de 94,5%.
Une fois l'essai Moderna terminé, le calcul final de l'efficacité du vaccin pourrait être inférieur, en raison de l'incertitude statistique.
Moderna a également présenté des preuves que son vaccin protège contre les cas graves de la COVID-19. L'analyse intermédiaire a révélé un total de 11 cas graves dans le groupe placebo de l'essai, et aucun dans le groupe vaccin.
Dans ses directives pour l'approbation d'urgence des vaccins anti-COVID, la FDA (Food and Drug Administration) américaine a déclaré que les essais d'efficacité devraient inclure au moins cinq cas graves dans le groupe placebo si un vaccin doit obtenir le feu vert. Pour plus d'information sur cet essai, voir ici.
Pour le moment on ne sait pas exactement combien de temps durent les effets protecteurs du vaccin, s'il peut empêcher les personnes contaminées de transmettre le virus ou s'il fonctionne aussi bien dans les groupes à haut risque comme les personnes âgées. La société a indiqué que, sur les 95 cas, 15 concernaient des personnes de plus de 65 ans, mais elle n'a pas précisé dans quelle branche de l'essai se trouvaient ces participants. Une question clé sera de savoir combien des 5 infections parmi les personnes vaccinées se sont produites chez des personnes de plus de 65 ans. Les chercheurs ont également été encouragés par le fait que ce vaccin reste stable dans les réfrigérateurs classiques pendant un mois et dans les congélateurs ordinaires pendant six mois, contrairement au vaccin de BioNtech-Pfizer qui doit être stocké à -70 °C avant d'être livré.
A suivre...
BNT162b1. 9 Novembre 2020.
Vaccin à ARN déjà présenté sur ce site (voir ci-dessous) les 1er et 21 Juillet 2020
BNT162b1 est un candidat vaccin à ARN (voir sur ce site : La course aux vaccins) développé par BioNtech, Mainz Allemagne et Pfizer, USA et Royaume Uni. L’ARN codant pour le domaine de liaison au récepteur de la protéine S (Spike) du SARS-CoV-2 est formulé dans des nanoparticules lipidiques et administré par voie intramusculaire en deux doses de 30 microgramme d'ARN par dose, séparées par 21 jours. Aujourd'hui, aucun vaccin à ARN n'avait encore été autorisé par les agences de régulation sanitaires aux Etats-Unis ou en Europe.
Les premières études de phase 1 et 2 étaient encourageantes (voir les rapports des 1er et 21 juillet ci-dessous), et les résultats de l'étude de phase 3 conduite sur 43 538 participants (aux Etats-Unis, Brésil, Argentine et Afrique du Sud) devraient être publiés courant Novembre 2020. Pour l'heure, 38 955 volontaires ont reçu la deuxième dose du vaccin. Je ne peux ici détailler le protocole de l'étude qui est très volunineux (à télécharger ici: ![]() Pfizer c4591001 clinical protocol nov2020 (1.31 Mo)), mais il est important de comprendre que parmi les 43 538 volontaires enrolés dans cette phase 3, certains pouvaient être positifs pour le SARS-CoV-2 à l'inclusion. L'efficacité du vaccin est évaluée sur la comparaison entre le nombre de participants infectés par le SARS-CoV-2 dans le bras "vaccin" par rapport au nombre de ceux infectés dans le bras "placebo".
Pfizer c4591001 clinical protocol nov2020 (1.31 Mo)), mais il est important de comprendre que parmi les 43 538 volontaires enrolés dans cette phase 3, certains pouvaient être positifs pour le SARS-CoV-2 à l'inclusion. L'efficacité du vaccin est évaluée sur la comparaison entre le nombre de participants infectés par le SARS-CoV-2 dans le bras "vaccin" par rapport au nombre de ceux infectés dans le bras "placebo".
Pour cette étude, Pfizer a fait appel à un groupe indépendant de chercheurs (le Conseil de Sécurité et de Surveillance des Données) pour examiner rapidement les données de l'étude avant son achèvement. Selon le rapport préliminaire émis par cette instance sur cette étude de phase 3, et comme largement commenté dans les médias depuis hier (9/11), les résultats semblent très prometteurs puisqu'ils montrent une efficacité du vaccin de l'ordre de 90%, sept jours après la deuxième dose, sur les patients participants et infectés par le SARS-CoV-2.
Toutefois dans l'état actuel d'avancement de l'étude, le nombre de ces patients infectés est encore faible (94) et les analyses à venir pourraient affecter les résultats. Par ailleurs, on ne sait pas encore si le vaccin prévient les cas qui peuvent entraîner une hospitalisation et un décès. De plus, il n'y a pas non plus d'informations à ce jour sur la question de savoir si le vaccin élimine le virus chez les porteurs asymptomatiques. Une autre question est de savoir quelle sera la durée pendant laquelle le vaccin restera efficace.
A suivre donc...
Virus complet inactivé. 15 Octobre 2020
Xia et al. Safety and immunogenicity of an inactivated SARS-CoV-2 vaccine, BBIBP-CorV: a randomised, double-blind, placebo-controlled, phase 1/2 trial. Lancet 2020.
Il s’agit d'un candidat vaccin (BBIBP-CorV) constitué par un virus SARS-CoV-2 (souche HB02) inactivé par traitement à la b-propriolactone. Ce vaccin a été développé et produit par un consortium chinois composé des institutions suivantes : 1. Beijing Institute of Biological Products Company Limited, Beijing, China. 2. National Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention (China CDC), Beijing, China. 3. National Animal Models for Human Diseases Resources Center, Beijing, China. 4. National Institute for Food and Drug Control, Beijing, China. 5. MOE Key Laboratory of Protein Science & Collaborative Innovation Center of Biotherapy, School of Medicine, Tsinghua University, Beijing, China. Ce vaccin a été étudié en préclinique sur divers modèles animaux (souris, cochon d’Inde, lapin, et primates, cynomologus et macaques rhesus). Chez ces espèces il induit des niveaux élevés d’anticorps neutralisants contre le SARS-CoV-2 et protège le macaque rhesus contre un réinfection par voie trachéale sans effet détectable délétère (Wang et al. Cell 2020).
Dans cette étude, les chercheurs ont évalué la sécurité sanitaire (non-toxicité/tolérance) et l'immunogénicité chez l'homme du BBIBP-CorV dans un essai randomisé, en double aveugle, contrôlé par placebo, de phase 1/2 a été entrepris à Shangqiu City, province du Henan, en Chine.
Dans la phase 1, 192 personnes en bonne santé âgées de 18 à 80 ans, séronégatives pour les anticorps IgM/IgG spécifiques du SRAS-CoV-2 au moment de l’essai ont été réparties au hasard en deux groupes d'âge (18-59 ans et ≥60 ans) pour recevoir le vaccin ou le placebo en deux doses de 2 μg, 4 μg, ou 8 μg aux jours 0 et 28.
Dans la phase 2, 448 adultes en bonne santé (âgés de 18-59 ans) ont été répartis au hasard par randomisation pour recevoir le vaccin (n=84 dans chaque groupe) ou le placebo (n=112) à une dose unique de 8 μg le jour 0, ou en deux doses de 4 μg les jours 0 et 14, 0 et 21, ou 0 et 28.
Les résultats principaux étaient la sécurité et la tolérance. Le résultat secondaire était l'immunogénicité, évaluée comme la neutraliser les réponses des anticorps contre le CoV-2 du SRAS infectieux. Cette étude est enregistrée auprès de www.chictr.org.cn, ChiCTR2000032459.
Résultats . Au moins un effet indésirable a été signalé dans les 7 premiers jours suivant l'inoculation du vaccin. L'effet indésirable systématique le plus fréquent était la fièvre. Tous les effets indésirables étaient de gravité légère ou modérée. Aucun effet indésirable grave n'a été signalé dans les 28 jours suivant la vaccination. Les titres moyens géométriques des anticorps neutralisants étaient plus élevés au jour 42 dans le groupe des 18-59 ans avec des valeurs de 87, 211 et 228 pour les doses 2, 4 et 8 μg, respectivement. Dans le groupe des 60 ans et plus ces valeurs étaient de 80, 131, et 170 pour les doses 2, 4 et 8 μg, respectivement.
Conclusion. Le vaccin BBIBP-CorV est sûr et bien toléré à toutes les doses testées dans les deux groupes d’âge des bénéficiaires. Au jour 42 après vaccination, les réponses humorales contre le SARS-CoV-2 ont été induites chez toutes les personnes vaccinées. Les titres des anticorps neutralisants les plus élevés ont été obtenus après vaccination en double dose aux jours 0 et 21 ou 28.
Adénovirus 26 et 5 recombinants. 4 Septembre 2020
Logunov et al. Safety and immunogenicity of an rAd26 and rAd5 vector-based heterologous prime-boost COVID-19 vaccine in two formulations: two open, non-randomised phase ½ studies from Russia Lancet 2020.
Cette étude concerne le vaccin russe appelé Sputnik, dont j’ai déjà parlé dans cette page « News sur les vaccins » (voir plus bas).
Les auteurs ont développé un vaccin anti-COVID-19 composé de deux éléments, un adénovirus recombinant de type 26 (rAd26) et un adénovirus recombinant de type 5 (rAd5), tous deux porteurs du gène de la protéine Skype (S) du SARS-CoV-2 (hCoV-19/Wuhan/Hu-1/2019), le virus responsable de la COVID-19 (rAd26-S et rAd5-S). Voir la page « La course aux vaccins » sur ce site.
Le but de l’étude était d’évaluer la sécurité sanitaire (non-toxicité/tolérance) et l'immunogénicité de deux formulations (congelée ou lyophilisée) de ce vaccin. Dans ce but, les chercheurs ont réalisé deux études ouvertes, non randomisées, de phase 1/2 dans deux hôpitaux : Burdenko Hospital and Sechenov University, Moscow, Russia. Des adultes volontaires en bonne santé (hommes et femmes) âgés de 18 à 60 ans ont été recrutés pour ces deux études. Dans la phase 1 de chaque étude, une dose de rAd26-S ou une dose de rAd5-S a été administrée par voie intramusculaire le jour 0, et la non-toxicité des deux éléments a été évaluée pendant 28 jours. Dans la phase 2 de l’étude, qui a commencé au plus tôt 5 jours après la phase 1 de la vaccination, le rAd26-S et le rAd5-S ont été administrés, respectivement le jour 0 et le jour 21, par voie intramusculaire selon une stratégie de vaccination en plusieurs étapes, qui permet de présenter un même antigène (la protéine S) à l'aide de différents vecteurs.
Les résultats principaux concernaient l'immunité humorale spécifique à l'antigène (anticorps spécifiques du SARS-CoV-2 mesurés par ELISA les jours 0, 14, 21, 28 et 42) et la non-toxicité (nombre de participants ayant subi des effets indésirables surveillés tout au long de l’étude). Les résultats secondaires concernaient l'immunité cellulaire spécifique à l'antigène (réponses des cellules T et interféron-γ) et les anticorps neutralisants (détectés par un test de neutralisation du SARS-CoV-2). Ces essais sont enregistrés auprès de ClinicalTrials.gov, NCT04436471 et NCT04437875.
Entre le 18 juin et le 3 août 2020, 76 participants ont été recrutés pour les deux études (38 dans chaque étude) : neuf volontaires ont reçu le rAd26-S lors de la phase 1, neuf ont reçu le rAd5-S lors de la phase 1, et 20 ont reçu le rAd26-S et le rAd5-S en phase 2. Les deux formulations du vaccin étaient sûres et bien tolérées. Les événements indésirables les plus fréquents étaient une douleur au point d'injection (44 [58%]), une hyperthermie (38 [50%]), des céphalées (32 [42%]), une asthénie (21 [28%]), et des douleurs musculaires et articulaires (18 [24%]). La plupart de ces effets indésirables étaient légers et aucun effet grave n'a été détecté. Tous les participants a produit des anticorps contre la glycoprotéine du SRAS-CoV-2. Au jour 42, les titres d'IgG spécifiques au domaine de liaison au récepteur étaient de 14 703 unités avec la formulation congelée et 11 143 unités avec la formulation lyophilisée, et les anticorps neutralisants étaient de 49,25 avec la formulation congelée et 45,95 avec la formulation lyophilisée, avec un taux de séroconversion de 100 %. Les réponses à médiation cellulaire ont été détectées chez tous les participants au 28e jour, avec une prolifération cellulaire médiane de 2 à 5 % de CD4+ et de 1 à 3 % de CD8+ avec la formulation congelée, et une prolifération cellulaire médiane de 1 à 3 % de CD4+ et de 1 à 1 % de CD8+ avec la formulation lyophilisée.
Le vaccin COVID-19 hétérologue à base de vecteurs rAd26 et rAd5 présente un bon profil de sécurité et induit de fortes réponses immunitaires humorales et cellulaires chez les participants. Il convient d'étudier plus avant l'efficacité de ce vaccin pour la prévention de la COVID-19 dans le cadre d’une phase 3 en cours (40 000 participants).
Virus complet inactivé. 13 Août 2020
Xia et al. Effect of an Inactivated Vaccine Against SARS-CoV-2 on Safety and Immunogenicity Outcomes. Interim Analysis of 2 Randomized Clinical Trials. JAMA2020
L’objectif de cette étude était d’évaluer la sécurité et l'immunogénicité d'un vaccin expérimental à virus entier (SARS-CoV-2 WIV04, inactivé par traitement à la beta-propionolactone) prélevé sur un malade à Wuhan (vaccin développé par Sinopharm et diverses universités chinoises). L'inactivation par la b-propionolactone est une technique déjà utilisée pour divers virus enveloppés dont le H3N2, produisant un virus incapable de pénétrer les cellules. Deux essais cliniques de phase 1 et 2 ont été randomisés en double aveugle et contrôlés par placebo, et conduits dans la province du Henan Chine, sur des adultes en bonne santé âgés de 18 à 59 ans, à partir du 12 avril 2020. L'analyse intermédiaire a été réalisée le 16 juin 2020 et mise à jour le 27 juillet 2020.
Dans l'essai de phase 1, 96 participants ont été répartis en 3 groupes de doses (2,5, 5 et 10 μg/dose) et un groupe d'adjuvants à base d'hydroxyde d'aluminium (alun) (n = 24 dans chaque groupe). Chaque participant a reçu 3 injections intramusculaires aux jours 0, 28 et 56.
Dans l’essai de phase 2, 224 adultes ont été répartis dans 2 groupes, chaque participant recevant une injection de 5 μg, les jours 0 et 14 pour le groupe 1 (n = 84) contre alun seulement (n = 28), et les jours 0 et 21 (n = 84) contre alun seulement (n = 28) pour le groupe 2.
Les 320 participants (âge moyen, 42,8 ans ; 200 femmes), ont tous terminé l'étude jusqu'à 28 jours après la vaccination complète. Le critère principal d’évaluation en matière de sécurité (phase 1) était l’observation des effets indésirables 7 jours après chaque injection, et le critère principal d’évaluation en matière d’immunogénicité (phase 2) était la production d’anticorps neutralisant 14 jours après la vaccination complète.
Dans l’essai de phase 1, des effets indésirables (douleur au point d’injection, fièvre légère, mais aucun effet sévère) sont survenus chez 3/24 participants du groupe placebo, 5/24 participants du groupe 2,5 μg/dose, 4/24 participants du groupe 5 μg/dose, et 6 :24 participants du groupe 10 μg/dose.
La moyenne géométrique des titres d'anticorps neutralisants dans les groupes 2,5, 5 et 10 μg/dose était de 316, 206 et 297, respectivement, chez les participants de l’essai de phase 1, et de 121 et 247 au 14e jour après la deuxième injection chez les participants de l’essai de phase 2 recevant le vaccin aux jours 0 et 14 et aux jours 0 et 21, respectivement. Aucune réponse anticorps n'a été détectée dans tous les groupes placebo.
En conclusion de cette étude intermédiaire pratiquée sur un vaccin à base de SARS-CoV-2 complet inactivé, les participants ont présenté un faible taux de réactions indésirables et les résultats ont montré une immunogénicité significative. Une confirmation finale de l’efficacité du vaccin et de ses effets indésirables à long terme nécessitera des essais de phase 3 (Identificateur du registre chinois des essais cliniques : ChiCTR2000031809).
SPOUTNIK-V. 10 Août 2020
C’est une annonce qui frappe le monde et qui rappelle les premières heures de la conquête spatiale. Le 10 Août 2020, le président Vladimir Poutine annonçait triomphalement que la Russie était parvenue à développer le premier vaccin efficace contre le Sars-Cov-2. « Ce matin, pour la première fois au monde, un vaccin contre le nouveau coronavirus a été enregistré (…) je sais qu'il est assez efficace, qu'il donne une immunité durable ». Pour témoigner de son efficacité, le président russe a ajouté que le vaccin avait passé avec succès l'intégralité des tests nécessaires, et qu'une de ses deux filles avait déjà été vaccinée. Une nouvelle qui soulèvera à coup sûr beaucoup d’interrogations dans les jours à venir. (CH, JIM).
Ce vaccin, vectorisé par adénovirus humain (Ad5) recombinant exprimant la protéine virale S, est similaire à celui développé par la compagnie chinoise CansinoBiologics (voir ci-dessous), Aucun résultat sur ce vaccin n'a encore été publié.
BNT162b1. 20 Juillet 2020
Sahin et al. Concurrent human antibody and TH1 type T-cell responses elicited by a COVID-19 RNA vaccine. MedRxiv 2020
Récemment, des données préliminaires sur le candidat vaccin BNT162b1, un ARN messager modifié codant pour le domaine de liaison au récepteur de la protéine S du SARS-CoV-2 formulé dans des nanoparticules lipidiques (voir sur ce site ICI) et développé par BioNtech, Mainz Allemagne et Pfizer, Hurley, Royaume Uni., ont été présentées sur cette page le 1er Juillet 2020 (voir plus bas sur cette page, Mulligan et al. MedRxiv 2020). La réponse immunitaire (anticorps et lymphocytes T cytotoxiques) après vaccination avec le BNT162b1 dans un deuxième essai ouvert non randomisé de phase 1/2 chez des adultes en bonne santé, âgés de 18 à 55 ans est présentée ici. Deux doses (1 et 50 μg) de BNT162b1 ont provoqué de fortes productions de cellules T cytotoxiques CD4+ et CD8+, et d’anticorps anti-protéine S avec des concentrations d'IgG nettement supérieures à celles présentes dans un panel de sérums humains de convalescence. La plupart des participants présentait des réponses immunitaires cellulaires (lymphocytes T cytotoxiques CD8+ et CD4+ spécifiques) de type TH1, avec production d’interféron γ dans une proportion élevée de ces cellules. Le vaccin BNT162b1 induit de multiples mécanismes bénéfiques susceptibles de protéger contre la COVID-19.
ChAdOx1 nCoV-19 ou AZD1222. 20 Juillet 2020
Folegatti et al. Safety and immunogenicity of the ChAdOx1 nCoV-19 vaccine against SARS-CoV-2: a preliminary report of a phase 1/2, single-blind, randomised controlled trial. Lancet 2020
Ces auteurs ont réalisé un essai contrôlé randomisé en simple aveugle de phase 1/2 sur cinq sites d'essai au Royaume-Uni avec le candidat vaccin ChAdOx1 nCoV-19 ou également connu sous la dénomination AZD1222 (voir sur ce site ICI). Il s'agit d'un adénovirus de Chimpanzé recombinant exprimant la protéine S du SARS-CoV-2, développé dans le cadre d’un consortium comprenant, l’Institut Jenner d’Oxford, l’Université d’Oxford, le BARDA (Biomedical Advanced Research and Development Authority), et AstraZeneca. Dans le groupe contrôle les personnes recevaient un vaccin anti-méningococcique conjugué (MenACWY). Des adultes en bonne santé, âgés de 18 à 55 ans ont été assignés au hasard (1:1) dans les groupes recevant le vaccin à une dose de 5 × 10¹⁰ particules virales ou le MenACWY en une seule injection intramusculaire. Un amendement au protocole dans deux des cinq sites a permis l'administration de paracétamol prophylactique avant la vaccination.
Entre le 23 avril et le 21 mai 2020, 1077 participants ont été inscrits et assignés à recevoir soit le ChAdOx1 nCoV-19 (n=543), soit le MenACWY (n=534). Les réactions locales et systémiques étaient plus fréquentes dans le groupe ChAdOx1 nCoV-19 et beaucoup ont été réduites par l'utilisation de paracétamol prophylactique, notamment les douleurs, sensations de fièvre, frissons, maux de tête, et malaise. Aucun événement indésirable grave lié au ChAdOx1 nCoV-19 n'a été constaté.
Le niveau des lymphocytes T cytotoxiques spécifiques de la protéine S du SARS-CoV-2 a atteint un maximum au 14e jour après vaccination. Le niveau des IgG anti-protéine S a augmenté au 28e jour et a été renforcés après une deuxième dose de vaccin. Les anticorps neutralisants contre le SARS-CoV-2 ont été détectés chez 91 à 100% des personnes vaccinées. Après une dose de rappel, tous les participants présentaient des anticorps neutralisants.
Ces résultats montrent que le ChAdOx1 nCoV-19 présente un profil de sécurité acceptable et permet l'induction de réponses immunitaires humorales (IgG) et cellulaires (lymphocytes T) satisfaisantes. Un programme de phase 3 est en cours (ClinicalTrials.gov, NCT04324606).
Ad5-nCoV. 20 Juillet 2020
Zhu et al. Safety, tolerability, and immunogenicity of a recombinant adenovirus type-5 vectored COVID-19 vaccine: a dose-escalation, open-label, non-randomised, first-in-human trial. Lancet 2020
Cette étude fait suite à celle publiée sur cette page le 22 Mai 2020.
Cet essai de phase 2 randomisé en double aveugle et contrôlé par placebo du vaccin anti-COVID-19, Ad5-nCoV un adénovirus 5 humain recombinant exprimant la protéine S du SARS-CoV-2, développé par l'Institut de Biotechnologie de Pékin et CanSino Biologics Inc. (voir sur ce site ICI) a été réalisé dans un seul centre à Wuhan Chine, afin de déterminer son efficacité en dose unique. Des adultes en bonne santé, âgés de 18 ans ou plus, séronégatifs pour le SARS-CoV-2, ont été sélectionnés pour cet essai et ont été répartis au hasard pour recevoir une seule injection du vaccin en intramusculaire dans le bras, à une dose de 10¹¹ particules virales par ml, ou 5 × 10¹⁰ particules virales par ml, ou le placebo.
Entre le 11 et le 16 avril 2020, 508 volontaires éligibles ont été recrutés (50% d'hommes ; âge moyen 39,7 ans) et ont accepté de participer à l'essai : 253 recevant le vaccin à la dose de 10¹¹ particules virales, 129 recevant le vaccin à la dose de 5 × 10¹⁰ particules virales, et 126 recevant le placebo.
Dans les deux groupes recevant le vaccin, le niveau des anticorps spécifiques à la protéine S (son domaine de liaison au récepteur) a atteint un pic au 28eme jour avec un taux de séroconversion de 96 % à 97 %. Les deux doses du vaccin ont induit des réponses significatives d'anticorps neutralisants et la production d’interféron γ chez 90 % (dose 10¹¹) et 88% (dose 5 × 10¹⁰) des personnes vaccinées.
Des effets indésirables ont été signalés chez 72 à 74 % des personnes vaccinées, mais aucune réaction indésirable grave n'a été documentée.
Ce vaccin est sûr et induit des réponses immunitaires importantes dans la majorité des cas après une seule immunisation (ClinicalTrials.gov, NCT04341389).
ARNm-1273. 14 Juillet 2020
Jackson et al. An mRNA Vaccine against SARS-CoV-2 - Preliminary Report. New England J Med 2020
ARNm-1273 est un ARN messager modifié, encapsulé dans des nanoparticules lipidiques exprimant la protéine S du SARS-CoV-2 développé par le National Institut des allergies et des maladies infectieuses (NIAID) en collaboration avec la firme américaine Moderna (voir sur ce site ICI).
Dans un essai ouvert de phase 1 à doses croissantes, 45 adultes en bonne santé de 18 à 55 ans ont reçu deux administrations à 28 jours d'intervalle, avec l'ARNm-1273 à la dose de 25 μg, 100 μg, ou 250 μg. Chaque groupe de doses comptait 15 participants.
Après la première vaccination, le taux des anticorps anti-protéine S augmentait avec la dose de vaccin administrée, et après la deuxième vaccination, les taux avaient encore augmenté de même que la présence d'anticorps neutralisants atteignant des niveaux similaires à ceux obtenues avec un panel d'échantillons de sérums de convalescence.
Les événements indésirables qui se sont produits chez plus de la moitié des participants, étaient : fatigue, frissons, maux de tête, myalgie, et douleur au point d'injection. Ces effets indésirables étaient plus fréquents après la deuxième vaccination, en particulier avec la dose la plus élevée, et trois participants (21 %) dans le groupe de dose 250 μg ont signalé un ou plusieurs événements indésirables graves.
Le vaccin ARNm-1273 induit des réponses immunitaires anti-SARS-CoV-2 chez tous les participants, et aucun problème de sécurité limitant les essais n'a été identifié. Ces conclusions encouragent à la poursuite du développement de ce vaccin (ClinicalTrials.gov, NCT04283461).
BNT162b1. 1 Juillet 2020
Mulligan et al. Phase 1/2 Study to Describe the Safety and Immunogenicity of a COVID-19 RNA Vaccine Candidate (BNT162b1) in Adults 18 to 55 Years of Age: Interim Report. MedRxiv 2020
BNT162b1 est un candidat vaccin à ARN (voir sur ce site : La course aux vaccins) développé par BioNtech, Mainz Allemagne et Pfizer, Hurley, Royaume Uni. L’ARN codant pour le domaine de liaison au récepteur de la protéine S (Skype) du SARS-CoV-2 est formulé dans des nanoparticules lipidiques et administré par voie intramusculaire.
Dans un article posté sur la plateforme MedRxiv le 1er Juillet dernier, les auteurs présentent une étude de phase 1/2, randomisée, contrôlée par placebo, en double aveugle sur la tolérance et l’immunogénicité du BNT162b1 chez 45 adultes volontaires (hommes et femmes) en bonne santé, âgés de 18 à 55 ans. Quatre groupes de volontaires ont été étudiés : 12 traités aux jours 1 et 21 par BNT162b1 à la dose de 10 μg, 12 traités aux jours 1 et 21 à la dose de 30 μg, 12 traités au jour 1 uniquement à la dose de 100 μg et 9 recevant le placebo. Les résultats ont été comparés à ceux obtenus sur un panel de sérums humains de convalescence (issus de patients du COVID-19 ayant survécu à l’infection).
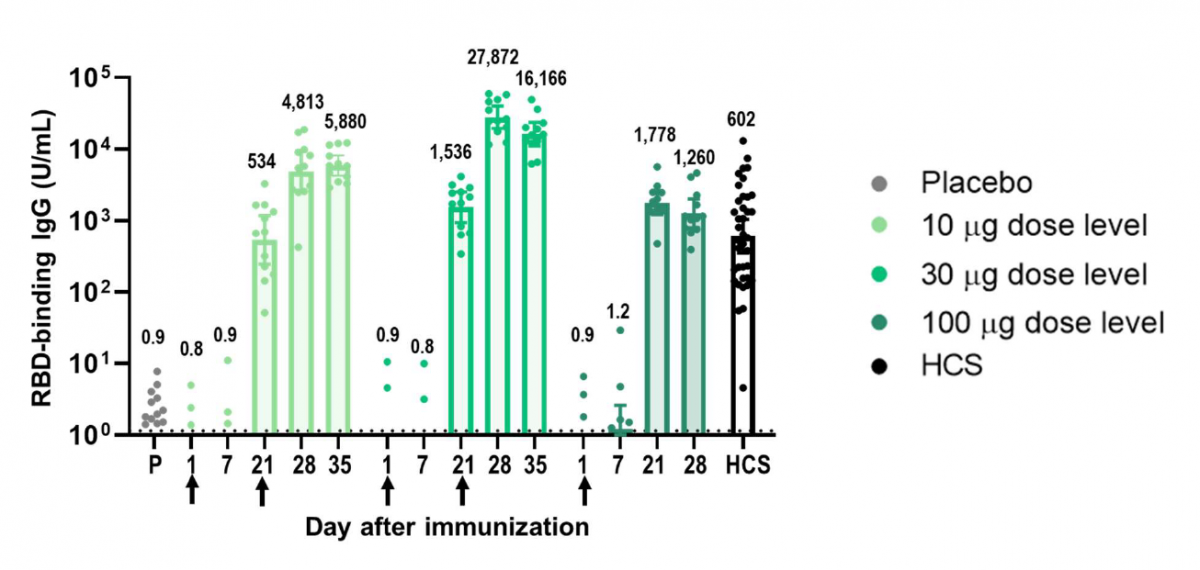
Niveau des IgG (anticorps) dirigés contre le domaine de liaison de la protéine S du SARS-CoV-2 au récepteur en fonction du temps du jour 1 (début du traitement) aux jours 7, 21, 28 et 35 après début du traitement (échelle logarithmique).
P : niveau des anticorps chez les volontaires traités par le placebo. HCS : niveau des anticorps dans le panel des sérums de convalescents. Les nombres figurant au-dessus des différentes barres du diagramme indiquent le niveau moyen des IgG chez les volontaires traités par le vaccin, par le placebo (P) ou dans lérum de convascents (HCS). Les flèches noires indiquent les jours où le vaccin a été administré. On peut voir le niveau élevé des IgG, notamment chez les personnes traitées avec la dose de 30 μg au jour 28 (27872 contre 602 chez les convalescents).
Mulligan et al. MedRxiv 2020.
Les réactions locales et les événements systémiques, généralement légers à modérés, et transitoires, ont été proportionnels à la dose de vaccin administrée. Une et deux semaines après la deuxième administration du vaccin (J28 et J35 ; doses 10 et 30 μg) ou après l’administration de la dose unique de 100 μg, le niveau des anticorps neutralisants était supérieur de 1,8 à 2,8 fois à celui observé dans un panel de sérums humains convalescents du COVID-19, tandis que le niveau des IgG (anticorps) était de 8 à 50 fois supérieur à celui observé dans ce même panel de sérums humains convalescents. Ces résultats très encourageants conduisent à poursuivre l’évaluation de ce candidat vaccin à ARNm à une plus large population.
Ad5-nCoV. 22 Mai 2020
Zhu et al. Safety, tolerability, and immunogenicity of a recombinant adenovirus type-5 vectored COVID-19 vaccine: a dose-escalation, open-label, non-randomised, first-in-human trial. The Lancet 2020.
Un vaccin à adénovirus a été développé dans le cadre du programme national de R&D de la Chine, du grand projet national de science et de technologie par CanSino Biologics. Il s’agit d’un adénovirus recombinant humain de type 5 (Ad5) exprimant la protéine S (Spyke) d'une souche de SARS-CoV-2 (voir sur ce site : La course aux vaccins).
Les auteurs publient un essai de phase 1 à doses croissantes, monocentrique, ouvert, non randomisé, sur 108 adultes de 18 à 60 ans en bonne santé, réalisé à Wuhan, Chine. Les volontaires (51% d’hommes) ont été répartis en 3 groupes de 36 recevant chacun une dose de vaccin (5 × 10¹⁰, 1 × 10¹¹ ou 1,5 × 10¹¹ particules virales) par voie intramusculaire. Le critère principal d’évaluation était l’observation des effets indésirables évalués entre 7 et 28 jours après vaccination. Les anticorps spécifiques ont été mesurés par ELISA, et les réponses des anticorps neutralisants induits par la vaccination ont été détectés à l'aide de tests de neutralisation du virus. Les réponses des lymphocytes T cytotoxiques ont été évaluées par des tests immuno-enzymatiques et par cytométrie de flux. Cette étude a été enregistrée sur le ClinicalTrials.gov, code NCT04313127.
La plupart des effets indésirables signalés (douleur, fièvre, fatigue, maux de tête) dans les différents groupes (entre 75 et 83%) étaient légers ou de gravité modérée. Aucun événement indésirable grave n'a été constaté dans les 28 jours suivant la vaccination. Les anticorps contre la protéine virale S (domaine de liaison au récepteur) et les anticorps neutralisants ont augmenté de manière significative au 14e jour, et ont atteint un pic 28 jours après vaccination. Le niveau des lymphocytes T cytotoxiques spécifiques CD4+ et CD8+ a atteint son point culminant au 14e jour après vaccination.
En conclusion, ce vaccin anti-COVID-19 vectorisé par l'Ad5 est tolérable et immunogène 28 jours après vaccination et les résultats justifient une étude plus approfondie.
Date de dernière mise à jour : 03/09/2021
Commentaires
-
Réponse à la question de Yves-Marie Beauverger
Vous trouverez la réponse à votre question sur la page: "Les candidats vaccins Français" sur ce site. -
Merci .En tant que médecin je trouve ces publications ( enfin un peu de rigueur scientifique à la place de discussions de café )très intéressantes . Ou en est Sanofi ? ( vaccin classique à virus atténué d'après ce que je crois ? )
-
trés bien expliqué. Mais j'ai un peu de mal à savoir pour lequel je pencherai...pour toi, ils se valent tous?
merci pour ces infos -
 Excellent résumé, et merci pour ces informations qui restent hors de notre portée. Il semble donc que doucement des solutions
Excellent résumé, et merci pour ces informations qui restent hors de notre portée. Il semble donc que doucement des solutions
se dessinent.



Ajouter un commentaire