
Syndrome de Sanfilippo
Nous avons déjà vu à maintes reprises les conséquences d’une grève des éboueurs dans certaines grandes villes. D’immenses tas d’ordures s’accumulent dans les rues et il est facile d’imaginer que si la grève se prolongeait pendant plusieurs années, la vie dans ces villes deviendrait absolument impossible.

Figure 1
C’est un peu se qui se passe dans le syndrome de Sanfilippo.
Syndrome de Sanfilippo
Encore appelé mucopolysaccharidose de type III, ce syndrome fait partie des maladies dites “de surcharge lysosomales”, un groupe d’une cinquantaine de maladies génétiques rares.
Ce syndrome a été récemment largement médiatisé avec le cas de Emma et Hugo, deux enfants de 21 et 36 mois et issus d’un même couple. Tous deux sont atteints du syndrome de Sanfilippo de type B. Rappelons que Hugo n’a présenté les premiers signes de la maladie qu’après la naissance d’Emma.

Figure 2
La fréquence de ce syndrome est très variable selon les pays. Sur 100 000 naissances, il touche en moyenne 1,9 naissance aux Pays-Bas, 1,7 en Australie, 1,2 au Royaume-Unis, 0,8 au Portugal, 0,7 en France et au Japon.
Les premiers symptômes apparaissent généralement entre 1 et 4 ans et le plus marquant d’entre eux est une neurodégénérescence sévère souvent accompagnée de dilatation ventriculaire, d’hydrocéphalie et d’épilepsie. Les signes les plus fréquents incluent : retard de développement mental, cognitif et physique avec perte des acquis, hyperactivité, comportement agressif, troubles du sommeil avec parfois inversion du rythme jour-nuit. On note également des troubles à d’autre niveaux, morphologiques, infections ORL, surdité, autonomie (perte d’initiative), osseux, respiratoires et gastrointestinaux (hépatomégalie). La survie moyenne des patients est de l’ordre de 13 à 15 ans.
Outre l’observation du tableau clinique ci-dessus, des explorations radiologiques et l’analyse de la présence de glycosaminoglycanes dans les urines et le sang, puis la mesure dans le sang des activités des enzymes lysosomales permet de poser le diagnostic d’un syndrome de Sanfilippo. Ce n’est que l’analyse génétique des mutations éventuelles sur les gènes SGSH, NAGLU, HGSNAT ou GSN qui permet de préciser le type du syndrome en A, B, C ou D (voir plus loin).
Maladies lysosomales
Les protéoglycanes
Les protéoglycanes sont des complexes qui se forment entre diverses protéines et des glycosaminoglycanes (aussi appelés mucopolysaccharides). Les glycosaminoglycanes sont des polymères saccharidiques (sucres modifiés) linéaires à longue chaîne. Il existe un grand nombre de glycosaminoglycanes comme les sulfates d’héparane, de chondroïtine, de dermatane, de kératane, et l’acide hyaluronique. Ils diffèrent par la nature des sucres modifiés et par leur longueur. Ils se fixent sur de nombreuses protéines, un peu comme des poils, pour en moduler l’activité biologique (voir le schéma ci-dessous). Les protéoglycanes remplissent des fonctions essentielles dans l’organisme. Par exemple, ils permettent de lubrifier les articulations (cartilages) ou divers canaux (mucus intestinal, respiratoire, etc.), de consolider divers organes (tissus conjonctifs), de favoriser les interactions et communications cellulaires (récepteurs cellulaires). Le glycosaminoglycane impliqué dans le syndrome de Sanfilippo est le sulfate d’héparane.
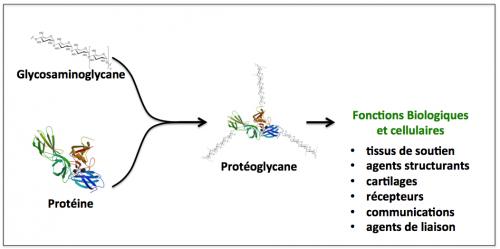
Figure 3
Lysosomes et dégradation des protéoglycanes
Comme toutes les protéines, les protéoglycanes sont synthétisés dans les cellules, ils remplissent leurs fonctions biologiques, puis ils sont dégradés pour être éliminés et recyclés. Cette dégradation qui met en jeu plusieurs enzymes s’effectue dans les lysosomes (voir ci-dessous).
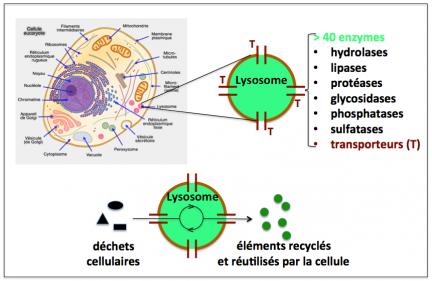
Figure 4
Les lysosomes sont de petites organelles cellulaires (environ 0,5 micron de diamètre) qui concentrent les déchets cellulaires et autres molécules destinés à être recyclés, grâce à des protéines de transport. Une fois à l’intérieur des lysosomes et du fait du milieu acide qui y règne, ces déchets sont biotransformés et dégradés par toute une batterie d’enzymes (on en compte au moins une quarantaine) incluant des hydrolases, lipases, protéases, etc. Les éléments biologiques issus de ces biotransformations (acides aminés, lipides, nucléosides, etc.) sont relargués dans la cellule et sont réutilisés pour la synthèse de nouvelles macromolécules (protéines, polymères, acides nucléiques, etc.).
On voit donc que les lysosomes agissent comme des centres de ramassage, de traitement et d’élimination des déchets, à l’instar des éboueurs, décharges, incinérateurs et stations d’épuration de nos villes.
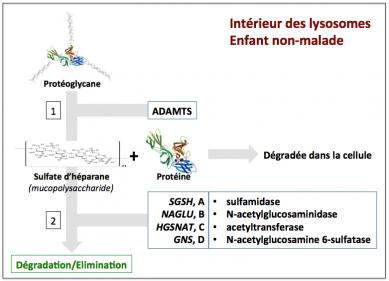
Figure 5
La figure ci-dessus montre comment s’opère la dégradation des protéoglycanes au sein des lysosomes chez l’enfant non-malade. J’ai choisi de montrer ici la dégradation du sulfate d’héparane, le glycosaminoglycane impliqué dans le syndrome de Sanfilippo de type B. Cette dégradation se fait en deux étapes. Dans l’étape 1, le protéoglycane est scindé en deux parties (protéine et sulfate d’héparane) par une enzyme appelée ADAMTS. La partie protéique est dégradée dans la cellule. Dans l’étape 2, la partie sulfate d’héparane est dégradée sous l’action d’une chaîne de réactions lysosomales comportant quatre sous-étapes (A, B, C, D) faisant intervenir quatre enzymes codées par les gènes SGSH, NAGLU, HGSNAT et GNS. C’est à dire que le produit de dégradation généré par la première enzyme (sous-étape 2A, sulfamidase) est repris par la deuxième (sous-étape 2B, N-acetylglucoronidase) et ainsi de suite. Au final de cette chaîne de réactions, le sulfate d’héparane est totalement dégradé et ses éléments sont recyclés. Si une des réactions de l’étape 2 n’a pas lieu, du fait d’une mutation dans le gène qui code pour l’enzyme en question, la dégradation des sulfates d’héparane stoppe et cela conduit à l’accumulation des déchets et à la maladie lysosomale.
Que s’est-il passé chez Emma et Hugo ?
Une grève, au sein des lysosomes…
Chacun de nous possède deux copies de tous les gènes présents dans le génome : une copie provient de la mère, une du père. Dans le cas de Emma et Hugo, les deux parents sont porteurs (mais non malades) d’une mutation dans l’une des deux copies de leur gène NAGLU. Ce gène code pour l’enzyme N-acétyglucosaminidase (NAGLU) responsable de la sous-étape 2B de la dégradation du sulfate d’héparane. Cette mutation, dont les parents évidemment ignoraient être porteurs, se traduit par l’absence ou l’inactivité de la protéine NAGLU. Emma et Hugo ont eu la malchance de recevoir les deux copies mutées de leurs parents. Si, par exemple, ils n’avaient reçu que la copie mutée de leur mère et la copie normale de leur père (ou vice versa), ils ne seraient pas malades.
Comme le montre le schéma ci-dessous, chez ces enfants, du fait de la mutation (croix rouge) sur les deux copies de leur gène NAGLU, l’enzyme N-acétylglucosaminidase n’est pas présente ou non fonctionnelle. Elle ne peut donc assurer sa fonction (sous-étape 2B), et la dégradation des molécules de sulfate d’héparane est incomplète. De ce fait, ces molécules s’accumulent dans les cellules et entraînent leur dysfonctionnement et la maladie.
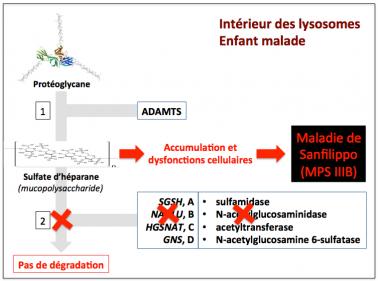
Figure 6
Comme ce phénomène se produit dans toutes les cellules de l’organisme, quasiment tous les tissus et organes sont touchés, y compris le cerveau. Nous savons aujourd’hui que chacun des quatre gènes impliqués dans la dégradation du sulfate d’héparane peut être muté, entraînant ainsi un syndrome de Sanfilippo (ou mucopolysaccharidose) de type A (si le gène muté est SGSH), B (NAGLU, le cas de Emma et Hugo), C (HGSNAT) ou D (GNS).
Il existe d’autres mucopolysaccharidoses liées à des mutations affectant d’autres gènes impliqués dans la dégradation d’autres mucopolysaccharides. D’où le grand nombre de maladies lysosomales.
Les stratégies thérapeutiques
Il n’existe aujourd’hui aucun traitement de ce syndrome.
Du fait que la manifestation la plus dramatique est le retard mental des enfants dès les premières années, les traitements en cours de développement sont ciblés sur le système nerveux central. Mais, traiter le cerveau n’est pas chose facile car cet organe est protégé par la barrière hémato encéphalique, un véritable rempart qui se charge de repousser tout intrusion de molécule étrangère. Ainsi, et sauf cas très particulier, tout administration de médicament par voie intraveineuse ou orale est vouée à l’échec. Diverses approches ont donc été testées en préclinique sur des modèles animaux :
- transplantation de cellules souches hématopoïétiques ou issues de sang de cordon ombilical : ces cellules passent la barrière hémato encéphalique et produisent les enzymes lysosomales manquantes
- remplacement de l’enzyme manquante ou non fonctionnelle par administration directe dans le liquide céphalorachidien ou dans le ventricule cérébral de l’enzyme normale que l’on sait synthétiser en laboratoire.
Bien que ces stratégies aient donné des résultats intéressants dans certaines maladies de surcharge lysosomale, elles se sont avérées décevantes ou encore à un stade trop prématuré de développement pour le cas du syndrome de Sanfilippo.
France : une première mondiale
Après diverses études précliniques sur des modèles animaux (rat et chien), l’équipe du Professeur Marc Tardieu* (CHU Le Kremlin-Bicêtre) vient de réaliser une première mondiale en traitant quatre enfants (âgés de 20, 26, 30 et 50 mois) atteints du syndrome de Sanfilippo de type B dans le cadre d’un essai clinique de phase I/II (vérification de la tolérance et de l’efficacité thérapeutique). Pour ce faire, les chercheurs ont introduit un fragment d’ADN codant pour l’enzyme N-acetylglucosaminidase (NAGLU) dans le génome d’un virus, le AVV2/5. Ils ont ensuite introduit ce virus AVV2/5-NAGLU directement dans le cerveau des enfants par microchirurgie (voir figure ci-dessous). Outre le fait que ce virus est inoffensif pour l’homme, il passe facilement la barrière hémato encéphalique et peut donc infecter des cellules du cerveau. Ce faisant, il délivre dans ces cellules l’ADN codant pour l’enzyme NAGLU manquante ou inactive. La chaîne des réactions de dégradation du sulfate d’héparane est ainsi rétablie, le niveau de sulfate d’héparane dans le sang et tout l’organisme diminue, et les symptômes régressent.
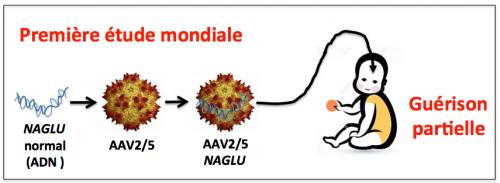
Figure 7
Les chercheurs ont vérifié que le virus AVV2/5 et l’enzyme NAGLU étaient bien présents dans le sang et le liquide céphalorachidien des enfants après traitement. Les enfants ont bien supporté ce traitement et leurs performances mesurées par une série de tests cognitifs, comportementaux et psychologiques adaptés, ont été significativement améliorées. Cependant, l’étude a montré que l’amélioration des symptômes était d’autant meilleure que l’âge des enfants était plus faible, suggérant ainsi un caractère irréversible ou difficilement réversible des lésions générées par la maladie au cours du développement cérébral. La conclusion de ce travail est que les enfants doivent être traités avant l’âge de 20 mois.
Ces premiers résultats très encourageants (ainsi que d’autres résultats obtenus sur d’autres maladies touchant les enfants) montrent que la thérapie génique peut être une solution dans le traitement de maladies génétiques rares.
Revenons maintenant au cas de Emma et Hugo
La famille a évidemment demandé à ce que ses enfants reçoivent un traitement similaire à celui décrit ci-dessus. Cela est possible, mais le problème en est le coût exorbitant. Plus de quatre millions d’Euros* ! D’où la forte médiatisation de ce cas sur les grandes chaînes de télévision nationales.
Quand on sait que la maladie de Sanfilippo de type B dont sont atteints Emma et Hugo ne touche en moyenne qu'une naissance par an en France, il paraît indécent que l'opération de ces deux enfants ne soit pas prise en charge par l'État. Oser demander 4 millions d'euros à une famille modeste pour pouvoir sauver ses deux enfants relève d'une absence totale d'éthique.
*Je me suis procuré le dossier budgétaire de cet essai clinique qui pourrait se tenir fin 2019. Les postes principaux concernent la production des lots de virus (1 250 000 €) et les études précliniques (850 000 €) sur les vérifications de non toxicité des virus.
Date de dernière mise à jour : 09/12/2018
Ajouter un commentaire