
Comment meurt-on de la Covid-19 ?
SARS-CoV-2, Covid-19 et orage cytokinique
Résumé tout public
Le SARS-CoV-2 entre le plus souvent dans notre organisme par les voies respiratoires et atteint très rapidement les bronches et les pneumocytes de type I et II des alvéoles pulmonaires.
-
Pourquoi le virus s’attaque-t-il précisément à ces cellules ?
Parce qu’elles présentent à leur surface une protéine appelée ACE2 qui constitue le récepteur du virus (sa porte d’entrée en quelque sorte). Toutes les cellules présentant ACE2 à leur surface sont susceptibles d’être infectées par le virus. Hormis les pneumocytes, c’est le cas des cellules de la muqueuse buccale, de la langue, de l’œsophage, de l’intestin grêle, du muscle cardiaque, du tube rénal, de la vessie, et d’autres vraisemblablement. Des troubles affectant ces organes sont effectivement rapportés en clinique chez les malades du Covid-19, mais ce sont surtout les troubles affectant le système respiratoire qui présentent la gravité la plus importante. En effet, dans le poumon, c’est au niveau des pneumocytes qu’ont lieu les échanges gazeux, oxygène passant des alvéoles vers le sang et dioxyde de carbone passant du sang vers les alvéoles. Le bon fonctionnement des pneumocytes est donc vital pour l’organisme.
-
Les systèmes de défense de l’organisme
Lors d’une infection virale (bactérienne, ou fongique), notre organisme réagit en mettant en place deux lignes de défense destinés à juguler l’infection : l’immunité innée qui agit de façon immédiate en attaquant le virus et les cellules infectées, et l’immunité adaptative qui produit une réponse plus tardive mais durable avec la production d’anticorps.
L’immunité innée débute par la reconnaissance de l’agent infectieux, ici le SARS-CoV-2, par des senseurs (ou capteurs) présents dans toutes nos cellules. Cet événement de reconnaissance conduit à l'activation d’un arsenal constitué d’interférons, de nombreuses protéines antivirales, de cytokines et chimiokines. Les cytokines et chimiokines agissent comme des messagers dont le but est de recruter sur le site de l’infection (en l’occurrence ici le poumon) les cellules de l’immunité innée comme les macrophages, neutrophiles, cellules tueuses, et autres. Ces cellules éliminent le virus de façon directe en le détruisant ou indirecte en détruisant les cellules qui en sont infectées et le produisent.
C’est alors que l’immunité adaptative se met en place. Son but est d’amplifier la réponse immunitaire innée contre l'agent infectieux par l’intermédiaire des lymphocytes T et B. Schématiquement, les lymphocytes T sont capables de tuer les cellules infectées et les lymphocytes B sécrètent dans le sang des anticorps dits « neutralisants » qui bloqueront toute nouvelle infection par le même virus.
-
Le virus peut-il contourner ces divers systèmes de défense ?
Hélas, oui ! Un grand nombre de pathogènes (virus de l’influenza H5N1, virus de l’hépatite C, et autres) et les coronavirus en particulier, sont capables d’inhiber plus ou moins fortement la réponse immunitaire de l’hôte, permettant et prolongeant ainsi leur survie. Dans les cas du SARS-CoV (pandémie de 2002-2003) et du MERS-CoV (pandémie de 2011), et vraisemblablement du SARS-CoV-2, le virus est capable d’inhiber fortement la réponse antivirale et de continuer à survivre et à se multiplier, notamment en infectant les cellules de la réponse innée (macrophages, neutrophiles et autres). Et ces cellules, une fois infectées, émettent des cytokines et chimiokines afin de recruter leurs congénères pour combattre l’infection. C’est la mise en place d’un cercle vicieux qui conduit à l’orage cytokinique.
-
Qu’est-ce qui provoque l’insuffisance respiratoire aigüe dans le Covid-19 ?
Dans un premier temps, plus le virus résiste aux cellules du système immunitaire inné, plus sa population augmente et plus le nombre de cellules infectées augmente, ce qui conduit à un recrutement massif des cellules immunitaires au niveau des alvéoles pulmonaires. Il s’en suit un emballement du système immunitaire inné pour détruire les cellules infectées. Cette situation hyper-inflammatoire qu’on appelle « l’orage cytokinique » conduit à l’attaque directe des cellules des alvéoles pulmonaires par les cellules du système immunitaire inné. La destruction de ces cellules entraîne la rupture physique des parois de alvéoles et des micro-vaisseaux sanguins qui les bordent. Il en résulte une « inondation » des alvéoles par le liquide interstitiel (lymphe) et le sang. Les échanges gazeux ne peuvent plus être réalisés correctement et le syndrome d’insuffisance respiratoire aigüe s’enclenche. Ce phénomène s’accompagne en général d’une diminution importante de la charge virale (faute de cellules au sein desquelles se multiplier). De fait, le malade meurt non pas du virus lui-même, mais de la suractivation de son système immunitaire inné. Un peu comme si pour pouvoir nettoyer une ville entièrement couverte de déchets, les éboueurs devaient détruire des bâtiments bordant les rues pour pouvoir intervenir.
Conclusion
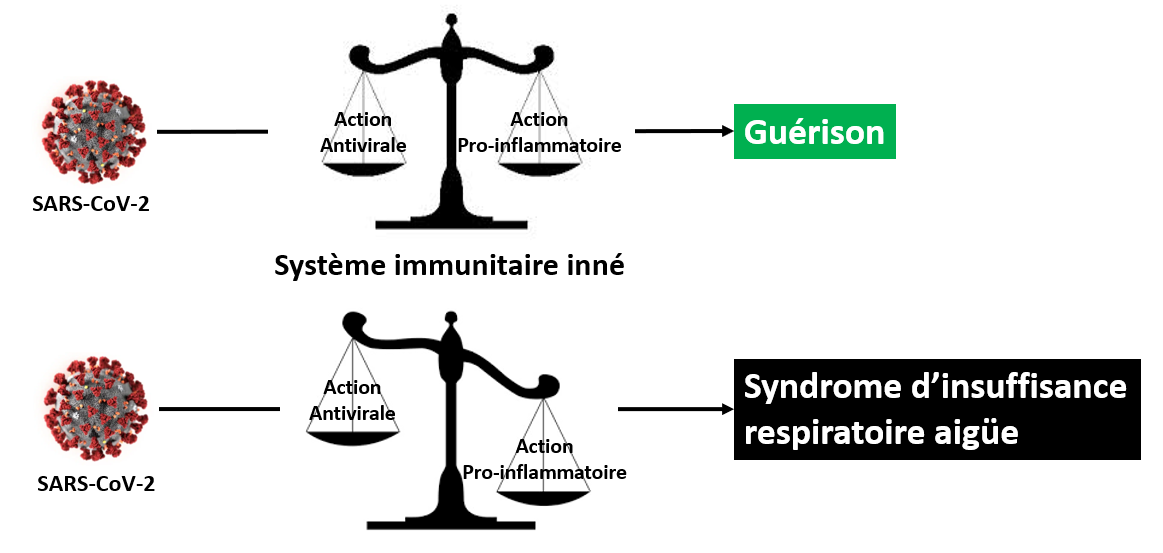
Dans le cadre d’une infection virale telle que celle amené par le SARS-CoV-2, la réponse immunitaire innée à un double caractère, antiviral et pro-inflammatoire, mettant en jeu des séries de gènes et protéines spécifiques dont le but est de juguler l’infection.
- L’action antivirale consiste à stopper la réplication du virus.
- L’action pro-inflammatoire consiste à mobiliser les cellules du système immunitaire afin de détruire les cellules infectées et les débris cellulaires engendrés sur le lieu de l’infection.
Lorsque l’équilibre entre ces deux actions, antivirale et pro-inflammatoire, est maintenu, la guérison est assurée.
Lorsque cet équilibre est rompu (par le virus, et du fait - vraisemblablement - de la génétique de l'hôte) au profit de l’action pro-inflammatoire, les pathologies extrêmes, comme le syndrome d'insuffisance respiratoire aigüe sévère, s’installent.
Et pendant ce temps-là, la chauve-souris se rit du coronavirus ! Pourquoi ?
En effet, pourquoi la chauve-souris survit-elle au coronavirus ? On sait que les chauves-souris constituent le réservoir naturel principal des coronavirus (et de nombreux autres virus) qui infectent l’homme, dont le SARS-CoV-2. Cependant, elles restent insensibles à ces infections.
Pourquoi ?
Des études récentes montrent que cette insensibilité serait due précisément au fait que chez ces animaux, l’action antivirale l’emporte toujours sur l’action pro-inflammatoire. En bref, l’inverse de ce qui se passe chez les patients dont l’état nécessite un passage dans une unité de soins intensifs.


Réponse immunitaire innée au SARS-CoV-2
Comparaison avec le SARS-CoV et le MERS-CoV
Retour sur les pandémies de SARS-CoV (2002-2003) et MERS-CoV (2011)
L'infection par le SARS-CoV entraînait une infection respiratoire qui allait de la maladie fébrile légère à un syndrome respiratoire aigu sévère (SARS) pouvant entraîner le décès (Peiris et al. Lancet 2003 ; Peiris et al. Nature Medicine 2004 ; Channappanavar et al. Seminar in Immunopathology 2017). L'évolution clinique se présentait en trois phases distinctes. La phase initiale était caractérisée par une réplication virale importante accompagnée de fièvre, toux et autres symptômes qui s’atténuaient en quelques jours. La deuxième phase était associée à une forte fièvre, une hypoxémie et une évolution vers des symptômes de type pneumonie, malgré un déclin progressif de la charge virale. Durant la troisième phase, environ 20% des patients progressaient vers le SARS et pour certains vers la mort (Nicholls et al. Respirology 2003 ; van den Brand et al. J Comp. Pathol 2014). En raison de la diminution progressive de la charge virale, la troisième phase était considérée comme résultant d’un emballement de la réaction inflammatoire de l'hôte. L’infection par le MERS-CoV (MERS, Middle East Respiratory Syndrome) entraînait les mêmes (sinon très voisins) symptômes avec de surcroit des troubles gastrointestinaux, douleurs abdominales, diarrhées et vomissements.
Covid-19, infection par le SARS-CoV-2
Fin 2019, l'apparition d'un autre coronavirus appelé le SARS-CoV-2 provoquant une pneumopathie voisine du SARS a été signalée à Wuhan, province de Hubei, en Chine. Cette maladie est désormais officiellement appelée "Covid-19". Le génome du SARS-CoV-2 a été entièrement séquencé (Wu et al. Nature 2020) et montre une séquence nucléotidique similaire mais distincte de celle du SARS-CoV et du MERS-CoV.
Grâce aux multiples études cliniques, au séquençage de nouvelle génération et au fait que les informations sont en libre accès sur le net, des données clés sur les caractéristiques cliniques des patients infectés et sur les réponses immunitaires de l'hôte commencent à apparaître (Huang et al. Lancet 2020). De plus, les similitudes de séquence avec SARS-CoV et MERS-CoV et les données expérimentales et cliniques obtenues sur ces deux coronavirus, permettent de faire des hypothèses sur la façon dont le système immunitaire de l'hôte va combattre le SARS-CoV-2 et comment celui-ci va tenter d’échapper aux réponses antivirales de l'hôte.
Comme dans les cas du SRAS et du MERS, les symptômes principaux les plus courants du Covid-19 sont la fièvre, la fatigue et les troubles respiratoires (toux sèche, mal de gorge et essoufflement). Certains patients qui développent une pneumopathie avec une opacité pulmonaire partielle ou totale dire « en verre dépoli » en imagerie par CT-scan, montrent une lymphopénie, une augmentation du taux des polynucléaires neutrophiles totaux et présentent des niveaux très élevés de nombreuses cytokines pro-inflammatoires (Wu et al. Nature 2020 ; Wang et al. Lancet 2020 ; Chan et al. Lancet 2020). Ces résultats sont conformes à ceux observés chez des patients atteints du SARS et du MERS et suggèrent un dérèglement de la réaction immunitaire de l’hôte avec une progression vers une pneumonie, un syndrome de détresse respiratoire aigu, une insuffisance respiratoire et potentiellement la mort. Cette issue fatale touche préférentiellement les personnes âgées et les personnes présentant des pathologies sous-jacentes telles que l'hypertension, les maladies cardiovasculaires, les cancers et le diabète.
Réponse immunitaire innée au SRAS-CoV-2 : comparaisons avec le SARS-CoV et le MERS-CoV
Les coronavirus sont des virus à ARN positif simple brin. Leur cycle de réplication est représenté ci-dessous.
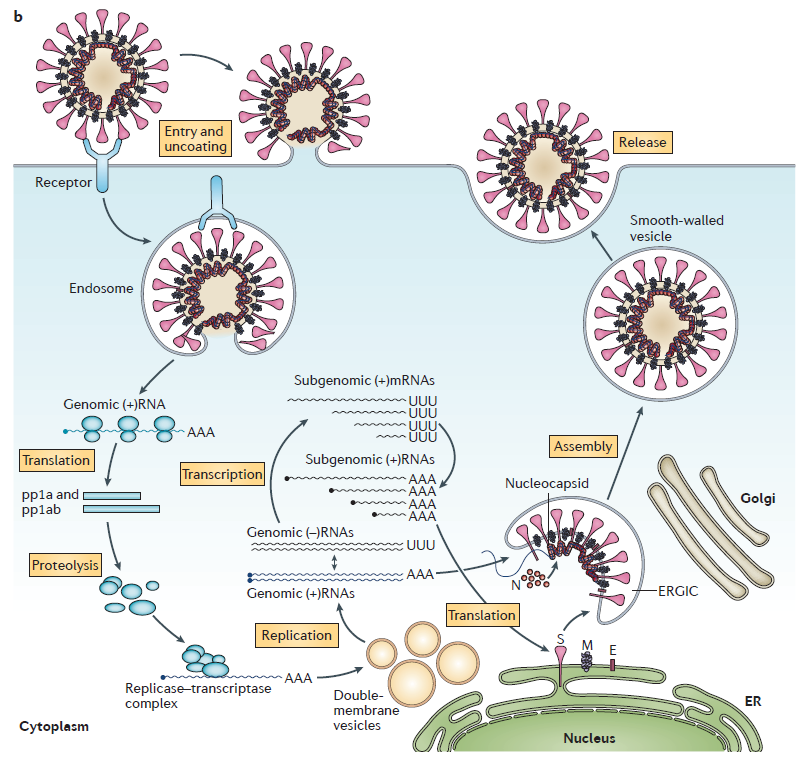
Cycle de réplication d'un coronavirus
(selon de Wit et al. Nature Review Microbiology 2016)
Comme le SRAS-CoV, le SARS-CoV-2 cible les cellules qui présentent à leur surface l’enzyme de conversion de l’angiotensine 2 (ACE2), et en particulier les cellules de l’appareil respiratoire situées au niveau des alvéoles pulmonaires qui assurent les échanges gazeux avec le sang. Ces cellules sont le pneumocytes de type I et II. Voir entrée du virus sur ce site ICI.
Première étape
Production de l’interféron (IFN)
Lors de l’infection d’une cellule par un virus, la réponse antivirale innée débute par la reconnaissance de l’agent infectieux grâce à un certain nombre de senseurs, les RPP (Receptor of Pathogen Pattern) tels que les TLR (Toll-Like Receptor) en particulier les TLR3 et TLR7, les senseurs d’ARN cytosolique RIG-1 et MAD5 (Prompetshara et al. APJAI 2020), les senseurs d’ADN cytosoliques notamment cGAS (cyclic GMP-AMP synthase) et les senseurs de dinucléotides comme STING (STimulator of INterferon Genes). Cette reconnaissance se fait sur la base de la détection d’ARN non protégé en position 5’ (les ARN de l’hôte sont protégés par une guanosine méthylée), ou d’ARN simple brin ou double brin (dans les intermédiaires de réplication du virus), ou enfin de la présence d’ADN dans le cytosol (dans les conditions normales, l’ADN est confiné dans le noyau et n'a aucune raison de se retrouver dans le cytosol). Ces divers événements de reconnaissance activent diverses voies de signalisation qui conduisent à l’activation de divers facteurs transcriptionnels comme IRF3, IRF7 et NF-κB, et de protéines impliquées dans ces voies de signalisation comme notamment STING et beaucoup d'autres. Une fois activés, IFR3 et IRF7 forment un dimère qui migre dans le noyau de la cellule et active l’expression du gène qui code pour l’interféron de type I (IFN-I).
Deuxième étape
Production des gènes stimulés par l’interféron (ISG)
L’IFN-I est sécrété par les cellules et se fixe sur son récepteur (IRNAR) présent sur la cellule émettrice ou sur les cellules avoisinantes. Après liaison sur son récepteur l'IFN active une voie de signalisation appelée JAK-STAT, dans laquelle les kinases JAK1 et TYK2 phosphorylent STAT1 et STAT2 (facteurs transcriptionnels). STAT1/2 forment un complexe avec IRF9, et migrent dans le noyau de la cellule pour activer la transcription des gènes stimulés par l'IFN (ISG). Ces gènes ISG codent pour des protéines antivirales dont la fonction est de détruire le virus. On estime à 200 à 500 le nombre de gènes ISG dans le génome humain. Par exemple, un des gènes ISG code pour la protéine PKR (Protéine Kinase R) qui bloque la synthèse des protéines et en particulier les protéines du virus. Une autre fonction de la PKR est de provoquer l’apoptose (mort cellulaire programmée) des cellules infectées par le virus. Un autre gène ISG code pour une protéine appelée RNAseL. Comme son nom l’indique RNAseL coupe les ARN messagers du virus. Un autre gène ISG code pour un protéine appelée MX1 qui bloque la réplication du génome viral. En bref, l'activité antivirale des ISG se produit à plusieurs niveaux : l'entrée du virus, la synthèse des ARN viraux codant pour les protéines virales, la synthèse des protéines virales, la disponibilité et la stabilité de l'ARN viral, la formation des particules virales, l’apoptose des cellules infectées et le renforcement général des réponses immunitaires innées et adaptatives.
Production de cytokines et chimiokines
NF-kB activé migre également dans le noyau et induit la production de cytokines et chimiokines pro-inflammatoires. Leur fonction est précisément de mobiliser les cellules du système immunitaire (globules blancs) sur le lieu de l'infection. Les chimiokines sont des cytokines chimiotactiques qui contrôlent la migration le positionnement au niveau des tissus lésés et l’activation des cellules du système immunitaire comme par exemple les cellules dendritiques, les macrophages, les monocytes, les neutrophiles polynucléaires. Ces cellules remplissent trois fonctions principales : phagocytose (digestion et destruction des agents pathogènes), destruction de cellules infectées, présentation des antigènes, production de cytokines (recrutement des cellules du système immunitaire).
A côté de la production de cytokines pro-inflammatoires en réponse à une infection virale par le facteur NF-kB, il existe une autre voie de production de ces cytokines, l’inflammasome NLRP3, un complexe multiprotéique présent dans le cytoplasme des cellules (Chen et al. Frontiers in Immunol 2019). L’inflammasome NLRP3 peut être activé en réponse à plusieurs types de stress, mais dans le cas d’une infection virale deux voies sont prépondérantes : i) activation par modification de flux ioniques (potassium et calcium) dus à certaines protéines virales, et ii) activation par les résidus de nucléotides libérés par la RNAseL sur l’ARN viral, via l’hélicase DHX33. Dès son activation, le NLRP3 s’associe aux protéines mitochondriales MAVS et aux protéines ACS qui recrutent la pro-caspase-1 pour former l'inflammasome NLRP3. Cet événement active la molécule en aval, la caspase-1, qui catalyse la production de l'interleukine 1b (l’IL-1b).
Les cellules sentinelles du système immunitaire inné (macrophages et monocytes) sont une source majeure de l'IL-1α et de l'IL-1β, mais de nombreux autres types de cellules épithéliales, endothéliales et fibroblastes, peuvent également produire ces interleukines. Après fixation sur son récepteur (IL-1R), IL-1b induit rapidement l'expression de l'ARN messager de centaines de gènes codant pour des cytokines pro-inflammatoire et autres protéines suivant diverses voies de signalisation impliquant de nombreux facteurs transcriptionnels et kinases. En parallèle, IL-1α et IL-1β induisent également l'expression de leurs propres gènes, ce qui fonctionne comme une boucle de rétroaction positive qui amplifie la réponse de l'IL-1 (Weber et al. Science Signaling 2010).
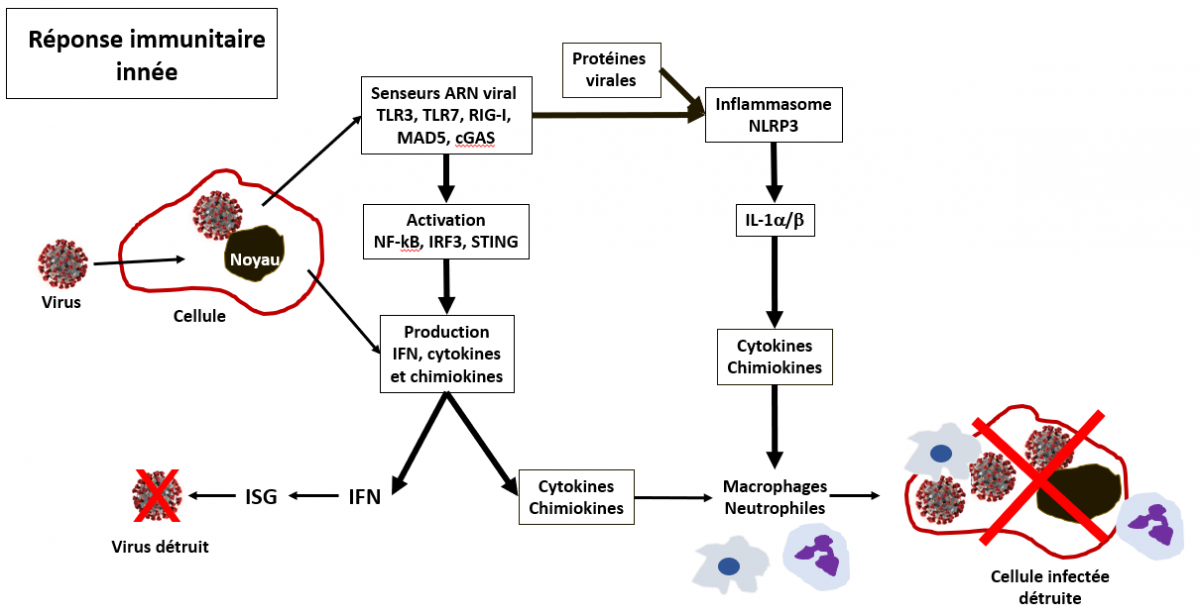
Schéma très simplifié de la réponse immunitaire innée
Seuls quelques uns des facteurs transcriptionnels et autres protéines de signalisation sont représentés ici.
L’ensemble de ces deux étapes, i) production d’interféron (IFN) et activation des ISG d’une part, et ii) production de cytokines et chimiokines d’autre part, avec le recrutement sur le lieu de l’attaque virale des macrophages, monocytes, polynucléaires neutrophiles et autres cellules tueuses constituent la première ligne de défense innée (Haller et al. Virology 2006 ; De Wit et al. Nat Rev Microbiol 2016) dont le but est de contrôler l'infection en diminuant ou inhibant la réplication du virus, et de détruire les cellules infectées.
Réponse immunitaire adaptative
L'immunité adaptative se met en place dès la reconnaissance de l’agent infectieux et son étiquetage comme « dangereux » par le système immunitaire inné en coordonnant et amplifiant la réponse immunitaire à l'agent infectieux par l’intermédiaire des lymphocytes T et B, permettant au final son élimination immédiate ou lors de nouvelles infections (Clarck et al. J Invest Dermatol 2005).
Contrairement aux cellules du système inné qui utilisent une répertoire de récepteurs fixes codés par le génome (TLRs, RIGs, MAD5), les cellules T et B subissent une recombinaison de gènes de récepteurs d'antigènes pour créer de nouveaux récepteurs d'antigènes uniques capables de reconnaître virtuellement tout antigène. Les cellules B et T qui ont rencontré l'antigène persistent à long terme dans un organisme et apportent des réponses rapides et spécifiques à la réinfection, un concept connue sous le nom de mémoire immunologique (Clarck et al. J Invest Dermatol 2005).
Réactions du virus à l’immunité innée
Dans de nombreux cas, le virus est éliminé plus ou moins rapidement selon qu’il est plus ou moins infectieux, plus ou moins toxique, et que l’hôte réagit plus ou moins efficacement et rapidement selon sa génétique, son âge, son état général, etc.
Cependant, un grand nombre de pathogènes, et en particulier les coronavirus et les virus de l’influenza (H5N1 et autres), sont capables de développer des stratégies leur permettant d’inhiber la réponse immunitaire de l’hôte, prolongeant ainsi leur survie.
Dans les cas de SARS-CoV (pandémie de 2002) et de MERS-CoV (pandémie de 2011), très largement étudiés depuis une quinzaine d’années, la réponse à l'infection virale par la production d'IFN de type I et la cascade aval aboutissant à l’expression des gènes de réponse à l’IFN (ISG) est fortement ou partiellement inhibée. Ce degré d’inhibition dépend probablement de plusieurs facteurs et il est possible que des polymorphismes génétiques propres à l’hôte soient responsable de cette variabilité de réponse. Cette stratégie d’atténuation de la réponse immunitaire de l’hôte par le virus est étroitement associée, et proportionnelle, à la gravité de la maladie (De Wit et al. Nat Rev Microbiol 2016 ; Channappanavar et al. Semin Immunopath 2017 ; Kindler et al. Adv Virus Res 2016). Potentiellement toutes les protéines virales, structurales et non structurales, participent à l’inhibition de la réponse immunitaire innée. Ces protéines sont capables par exemple de bloquer les voies de signalisation qui contrôlent la production de l’IFN et des autres cytokines pro-inflammatoires (interféron gamma par exemple) et la production des gènes ISG qui codent pour des protéines antivirales.
Le SARS-CoV-2 présente une similarité génomique globale avec le SARS-CoV ou MERS-CoV d’environ 79% et 50%, respectivement. En outre, les séquences d'acides aminés de certains protéines du SRAS-CoV-2 présentent 68% de similitude avec celles du SARS-CoV (Lu et al. Lancet 2020 ; Fung et al. Emerging Microbes & Infections 2020). Bien que le génome du SARS-CoV-2 contienne des gènes supplémentaires non présents dans SARS-CoV et MERS-CoV il est vraisemblable que le SRAS-CoV-2 utilise des stratégies similaires pour moduler la réponse immunitaire innée de l'hôte, notamment en ce qui concerne l'inhibition de la production d'IFN et de l’activation des ISG.
Par ailleurs, le SARS-CoV et le SRAS-CoV-2 utilisent le même récepteur d'entrée dans les cellules, l'ACE2 (angiotensin converting enzyme 2) (Voir sur ce site ICI). Le récepteur présumé du SARS-CoV-2, ACE2, est principalement exprimé dans un petit sous-ensemble de cellules dans le poumon, les pneumocytes alvéolaires de type II (Zhu et al. New England J Med 2020). Le SARS-Co-V infecte directement les macrophages et les cellules T, un élément clé de la pathogenèse médiée par ce virus (Perlman et al. Nat Rev Immunol. 2005). Pour l’heure, on ignore encore s’il en est de même pour le SARS-CoV-2 mais étant donné sa proximité génomique avec le SRAS-CoV il est vraisemblable qu’il puisse agir de la même façon, comme le suggèrent les données cliniques. Rappelons que les macrophages sont impliqués dans l’élimination des déchets cellulaires (débris de cellules et cellules nécrosées ou mortes) et que les polynucléaires neutrophiles, vitaux pour la défense de l'hôte, peuvent dans le cas d’une activation anormale de leur mobilisation entraîner des lésions tissulaires majeures par libération de chimiokines, cytokines cytotoxiques, protéases à sérine (élastase, protéinase 3, cathepsine G, dipeptidyl-peptidase I), métalloprotéases matricielles (MMP-2, -8, -9), polypeptides cationiques (lactoferrine, LL37) et les espèces réactives de l'oxygène (ROS) (Grommes et al. Mol Med 2011).
Orage cytokinique
Les informations cliniques sur le statut immunitaire inné des patients infectés par le SARS-CoV-2 montrent des tendances identiques, notamment pour ceux dont l’état nécessite le passage dans les unités de soins intensifs. Une étude réalisée sur 99 cas à Wuhan a montré une augmentation du taux des polynucléaires neutrophiles totaux (38%, cellules blanches du système immunitaire inné recrutés dans les tissus infectés par les cytokines pro-inflammatoires), une diminution des lymphocytes totaux (35%), et une augmentation de l'IL-6 sérique (52%, une cytokine pro-inflammatoire) et de la protéine c-réactive (84%, une protéine impliquée dans l’élimination des microorganismes, des cellules infectées et débris cellulaires) (Zhou et al. Nature 2020). Une autre étude également réalisée à Wuhan a montré chez 41 patients les mêmes résultats en particulier chez ceux placés en unités de soins intensifs par rapport aux autres patients atteints moins sévèrement ; ces modifications étaient statistiquement différentes. L’augmentation du nombre des polynucléaires neutrophiles et la diminution des lymphocytes serait en corrélation avec la gravité de la maladie et sa létalité (Wu et al. Nature 2020). En outre, les patients nécessitant un placement en unité de soins intensifs avaient des taux plasmatiques plus élevés de nombreuses cytokines de la réponse innée, IP-10, MCP-1, MIP-1A, et TNFα (Huang et al. Lancet 2020). Des niveaux élevés de cytokines pro-inflammatoires telles que le TNFα, l’IL-1b et l’IL-6 ont été détectés dans les autopsies de tissus infectés de patients atteints de SRAS (He et al., 2006). L’ensemble de ces caractéristiques cliniques suggèrent l'implication d'un état hautement pro-inflammatoire dans la progression et la gravité de la maladie qu’on appelle l’orage cytokinique.
Cette hausse précoce et massive des taux sériques de cytokines pro-inflammatoires et l’afflux important dans le tissu lésé des polynucléaires neutrophiles et des monocytes-macrophages avaient déjà été constamment observés dans les cas les plus sévères d’infection par le SRAS-CoV et le MERS-CoV (Mahallawi et al. Cytokine 2018 ; Wong et al. Clin Exp Immunol 2004 ; Perlman et al. Nat Rev Immunol. 2005 ; Zumla et al. Lancet 2015). Dans un modèle de souris infectées par le SARS-CoV, la dérégulation de la réponse à l’IFN et l’activité des monocytes-macrophages ont été identifiées comme la principale cause de pneumonie mortelle (Channappanavar et al. Semin Immunopath 2017). Une production excessive d’IFN et de cytokines pro-inflammatoires et de chimiokines et une mobilisation/infiltration importante de ces cellules au niveau du tissu pulmonaire par des polynucléaires neutrophiles hyper-inflammatoires et de monocytes-macrophages semblent être la cause principale des troubles pulmonaires sévères, c’est à dire le SARS.
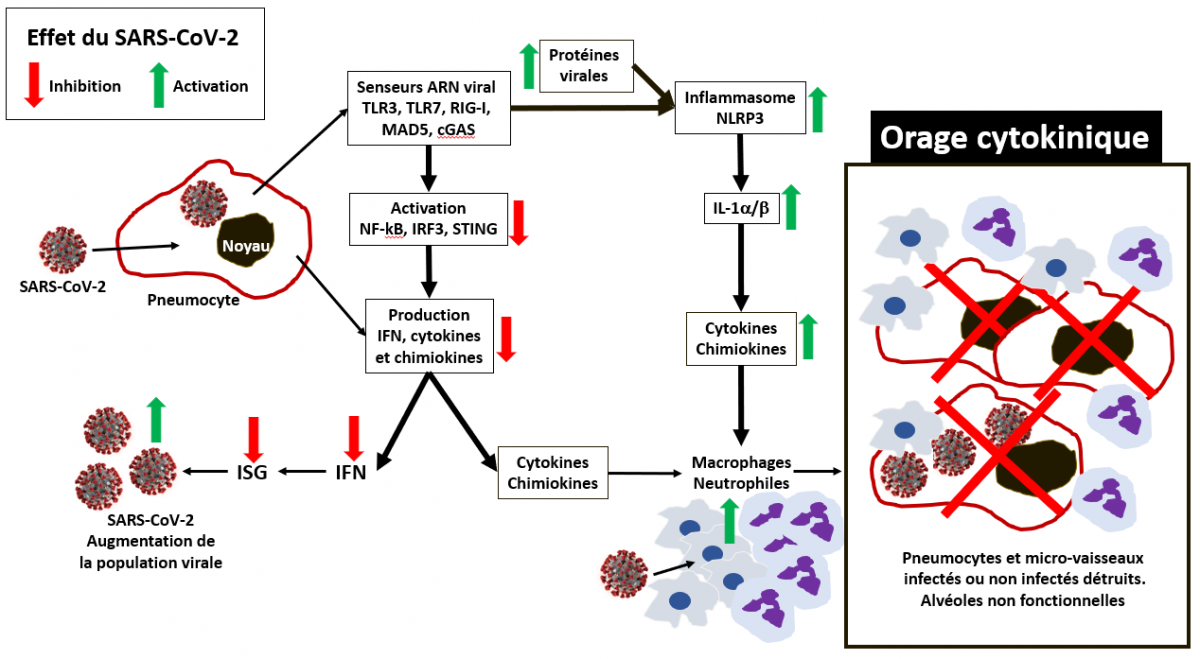
Schéma très simplifié de l'orage cytokinique
Les flêches vertes décrivent une augmentation et les flêches rouges une diminution des divers éléments en regard (virus, facteurs de transcriptions et autres protéines, cytokines, cellules du système immunitaire inné) résultant de l'effet modulateur du SARS-CoV-2 sur le système immunitaire inné.
On peut donc proposer le scénario suivant. Dans les infections dues au SRAS-CoV ou au MERS-CoV, le retard et/ou la forte atténuation de la mobilisation de l’IFN compromet fortement la destruction du virus dont la population progresse de façon massive et non contrôlée, entraînant un afflux massif de polynucléaires neutrophiles hyper-inflammatoires et de monocytes-macrophages vers les pneumocytes de type I et II, constituants majeurs des alvéoles pulmonaires. L'accumulation de ces cellules entraîne au mieux des affections respiratoires aiguës et au pire le syndrome aigu de détresse respiratoire et la mort.
Dans l'infection par le SRAS-CoV-2, un scénario similaire est attendu avec un degré variable d'interférence immunitaire. En effet il est intéressant de noter que la transmission du virus se produit même chez les personnes infectées asymptomatiques, suggérant une activation retardée de la réponse immunitaire innée. Avec des changements similaires dans les neutrophiles totaux et les lymphocytes pendant le Covid-19, le SARS-CoV-2 induit probablement un retard d’activation de l'IFN de type I et perte du contrôle viral dans une phase précoce d'infection. En inhibant la réponse immunitaire innée, le virus continue à survivre et à se multiplier, notamment en infectant les cellules de la réponse innée (macrophages, neutrophiles et autres). Et ces cellules, une fois infectées, émettent des cytokines et chimiokines afin de recruter leurs congénères pour combattre l’infection. C’est la mise en place d’un cercle vicieux. Il s’en suit un emballement du système immunitaire inné pour détruire les cellules infectées. Cette situation hyper-inflammatoire qu’on appelle « l’orage cytokinique » conduit à l’attaque directe des cellules des alvéoles pulmonaires infectées ou non infectées par les cellules du système immunitaire inné. La destruction de ces cellules entraîne la rupture physique des parois de alvéoles et des micro-vaisseaux sanguins qui les bordent. Il en résulte « l’inondation » des alvéoles par le liquide interstitiel (lymphe) et le sang. Les échanges gazeux ne peuvent plus être réalisés correctement et le syndrome d’insuffisance respiratoire aigüe s’enclenche. De fait, le malade meurt non pas du virus dont la charge virale montre très souvent un effondrement, mais de la suractivation de son système immunitaire inné.
Et pendant ce temps-là, la chauve-souris se rit du coronavirus !
Pourquoi ?
Seuls mammifères volants, les chauves-souris sont connues comme un réservoir de divers virus pathogènes pour l'homme, tels que le virus de la rage, les virus Nipah et Hendra, le virus Ebola, les virus de la grippe et les coronavirus. Ces animaux sont capables de survivre à des infections virales expérimentales in vivo avec ces virus à des doses létales chez d'autres mammifères (Rodhain, Bull. Soc. Pathol. Exot. 2015). Les mécanismes responsables de cette capacité des chauves-souris à coexister avec les virus sont encore mal connus, mais diverses études sur le sujet peuvent être résumées ici et apportent des éléments d’explication très intéressants (Fung et al. Emerging Microbes & Infections 2020):
- Le niveau élevé d'espèces réactives de l'oxygène (ROS) dû à l’activité métabolique importante des chauves-souris (le vol en particulier), peut entraîner une diminution de la réplication du CoV à un niveau gérable pour l’animal.
- La dégénérescence des senseurs d’agents pathogènes et de la voie de signalisation de NF-κB chez les chauves-souris atténue l’impact de l’infection virale (Zhang et al. Science 2013).
- L’expression constitutive (continuelle, non stimulée par le virus et autres pathogènes) des gènes qui codent pour les IFN de type I dans les tissus et les cellules de chauve-souris permet de juguler l’infection virale du fait d’un niveau constant de protéines antivirales codées par les gènes stimulés par l'IFN (ISG) (Zhou et al. Proc. Natl. Acad. Sci. 2016)
- La voie de signalisation STING est défectueuse chez les chauves-souris du fait de la présence d’une mutation unique (en S358) chez cette espèce par rapport aux mammifères dont l’homme (Xie et al. Cell Host Microbe 2018). De plus, l'activation de l’inflammasone NLRP3 est atténuée chez les chauves-souris (Ahn et al. Nature Microbiology 2019). Ces deux particularités pourraient contribuer à éviter la suractivation du système immunitaire inné lors d’infections virales.
En d’autres termes, il semble que chez les chauves-souris, les voies de signalisation qui contrôlent la réponse inflammatoire soient atténuées au profit de celles qui contrôlent la réponse antivirale.
La raison de cette situation pourrait résider dans le fait que les chauves-souris présentent un métabolisme oxydatif très élevé du fait du vol (comme dans le cas des oiseaux). Ce métabolisme oxydatif particulier est connu pour générer des ROS qui entraînent des atteintes multiples au niveau de l’ADN et des macromolécules cellulaires, autant d’évènements qui normalement enclenchent et entretiennent des réponses inflammatoires.
L’hypothèse est que les chauves-souris se sont adaptées à cette situation précisément en atténuant ces réponses inflammatoires liées à la production importante de ROS. Une première conséquence de cette adaptation serait l’extrême longévité de ces animaux (entre 20 et 30 ans) alors que la souris ne vit que 1 à 2 ans. Une autre conséquence est que cette adaptation a « profité » à la survie de ces animaux vis-à-vis des infections virales. Ces caractéristiques, associées à leur longue durée de vie, leur hibernation, leur comportement social favorisant les regroupements massifs entre individus, et leur nombre, font des chauve-souris de véritables laboratoires de virologie au sein desquels de nouveaux virus peuvent être produits en continue avec des possibilité multiples de recombinaisons.
Date de dernière mise à jour : 23/03/2026
Commentaires
-
-
Merci mille fois pour ces explications remarquables et en fracais .Cordialement Victoire chateau
-
 Merci Patrick pour cette vulgarisation sans laquelle il nous serait impossible de comprendre.
Merci Patrick pour cette vulgarisation sans laquelle il nous serait impossible de comprendre.


Ajouter un commentaire