
Syndrome de l'X fragile
Edition épigénétique par le système CRISPR-Cas comme approche thérapeutique dans le traitement du syndrome de l’X fragile
Résumé
- Le syndrome de l’X fragile est une maladie rare qui se caractérise par un déficit intellectuel léger à sévère, un QI compris entre 20 et 60, une hyperactivité, un déficit d’attention et des comportements proches de l’autisme. Elle peut s’accompagner de crises d’épilepsie, de troubles de la coordination motrice et de signes physiques comme une dysmorphie faciale. Cette maladie touche 1 garçon sur 5000 et 1 fille sur 8000 (la maladie étant atténuée chez ces dernières).
- Cette maladie est due à une mutation dans le gène FMR1 situé sur le chromosome X. Cette mutation rend le gène inactif, d'où la maladie.
- A l'heure actuelle il n'existe pas de traitement.
- Des chercheurs viennent d’utiliser une nouvelle méthode d’édition génomique et épigénomique (CRISPR-Cas9) permettant de corriger la mutation et ses effets. Cette approche thérapeutique pourrait être utilisée dans un futur proche pour traiter le syndrome de l’X fragile.
Le syndrome de l’X fragile
Le syndrome de l’X fragile est une des maladies génétiques rares les plus fréquentes avec une prévalence de 1 sur 5000. Elle se caractérise par un déficit intellectuel léger à sévère, un QI compris entre 20 et 60, une hyperactivité, un déficit d’attention et des comportements proches de l’autisme. De fait, 25% des malades atteints du syndrome de l’X fragile sont autistes. Ils ont également une propension élevée aux crises d’épilepsie et à des troubles de coordination motrice. Ce syndrome peut également être accompagné de signes physiques caractéristiques, comme la dysmorphie faciale, le prolapsus mitral, la macro orchidie (Sheridan 2011 ; Bhattacharyya 2013 ; Wijetunge 2013 ; Lozano 2014 ; Rajaratnam 2017).
Dans plus de 99% des cas, le syndrome de l'X fragile est due à une mutation dans le gène FMR1 (Fragile Mental Retardation 1) situé sur le chromosome X (q27.3) codant pour la protéine FMRP (Fragile Mental Retardation Protein). Cette mutation consiste en une séquence répétée du triplet CGG de longueur anormale (CGGn, avec n>230), et se situe dans la région 5’-non traduite du gène (Figure 1). C’est à dire la région comprise entre le site d’initiation de la transcription et le site de démarrage de la traduction (triplet ATG). La mutation n’affecte donc pas la séquence de la protéine mais affecte le niveau d’expression du gène et au final le niveau d’expression de la protéine. En effet, lorsque le nombre de CGG dans la séquence répétée est >230 (mutation dite “complète”), la région régulatrice (promoteur) de FMR1 est hyperméthylée sur les ilots CpG et donc inactivée. D’où l’absence d’ARNm (ARN messager) et de protéine FMRP chez les malades porteurs de cette séquence CGGn avec n>230. Diverses maladies génétiques résultent de la présence de séquences de triplets de nucléotides répétés anormalement longues comme par exemple la maladie de Huntington (voir sur ce site). On peut considérer que le syndrome de l’X fragile est un désordre d’origine épigénétique : ce n’est pas la séquence répétée CGG qui est responsable de la pathologie mais l’inhibition de la transcription du gène FMR1 qu’elle entraîne. Ceci est très différent du cas de la maladie de Huntington où c’est la séquence répétée (CAGn) qui est toxique.
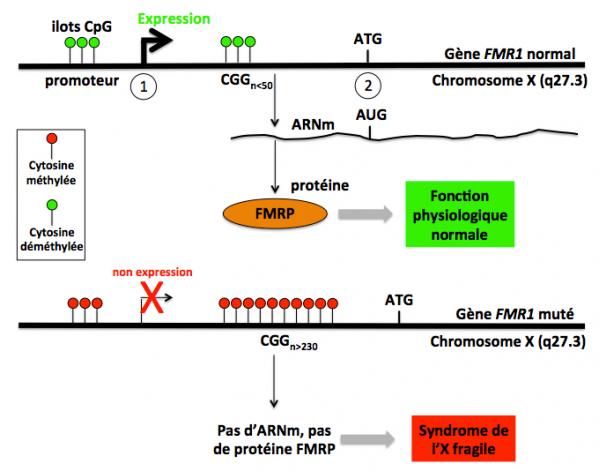
Figure 1: Le gène FMR1 normal et muté. Le trait noir horizontal représente le gène FMR1 situé sur le chromosome X en position q27.3. En 1 et 2, les sites d'initiation de la transcription (ARN) et de la traduction (protéine), respectivement. La séquence située en amont du site d'initiation de la transcription est le promoteur du gène, c'est à dire la région qui assure la modulation de son activité transcriptionnelle. Dans cette région, des séquences riches en C et G (ilots CpG) peuvent être méthylées sur les cytosines (C). Les ilots CpG non méthylés (cercles vert) favorisent l'expression du gène, alors que lorsque ces ilots sont hyperméthylés (cercles rouge) la transcription du gène est inhibée du fait de la modification de la chromatine. Partie haute de la figure: dans le gène FMR1 normal (région non codante), le nombre des triplets CGG répétés est variable selon les individus, mais inférieur à 50. Dans ces cas, les cytosines des triplets répétés et des ilots CpG ne sont pas méthylées et le gène est exprimé normalement avec production de l'ARNm, de la protéine et la fonction physiologique normale est assurée. Partie basse de la figure: chez les individus porteurs de la mutation dite “complète”, le nombre des triplets CGG répétés est supérieur à 230. Dans ces cas, les cytosines des triplets répétés et des ilots CpG sont hyperméthylées et l'expression du gène est nulle ou très faible, avec pour conséquence la mise en place du syndrome de l'X fragile.
FMRP est une protéine cytoplasmique qui se lie aux ARN. Elle est notamment impliquée dans le transport des ARN messagers dans les dendrites des neurones. On sait également qu’elle est liée aux ribosomes et régule la traduction des ARN messagers en protéines, une fonction importante pour le développement et la plasticité des synapses. En absence de FMRP, la synthèse protéique augmente avec pour conséquence une activation de diverses voies de signalisation intracellulaire. Des études animales montrent que FMRP est également impliquée dans le développement neuronal et qu’un déficit en cette protéine favorise la différenciation des cellules souches neurales en lignées gliales au dépend des neurones, entraînant ainsi une réduction de la production de neurones. Enfin, les neurones déficients en FMRP ne se développent pas normalement, notamment au niveau des dendrites.
Chez les individus normaux, la valeur de n (le nombre de triplet CGG dans le promoteur de FMR1) est généralement comprise entre 9 et 40. Lorsque n est compris entre 41 et 60 le génotype est dit “intermédiaire” et lorsqu’il est compris entre 50 et 200 il est dit en “prémutation”. Puisque le syndrome de l’X fragile est une maladie liée à l’X, sa fréquence est plus élevée chez les garçons que chez les filles (1/8000) qui, de plus, présentent un phénotype moins sévère du fait de la présence de deux chromosomes X (voir sur ce site : Dossier Maladies Rares: Les diverses causes).
En bref, lorsque le gène FMR1 comporte une séquence répétée de triplet CGG anormalement longue (>230), cette séquence ainsi que le promoteur du gène sont hyperméthylés et le gène n’est plus transcrit. Résultat, la protéine FMRP codée par ce gène n’est plus exprimée et le syndrome de l’X fragile s’installe. On peut donc imaginer deux alternatives qui permettraient de rétablir la transcription du gène et guérir la maladie: i) déméthyler les cytosines (C) de la séquence CGGn et du promoter, ii) raccourcir la séquence CGGn anormalement longue.
C’est ce qui a été fait dans le travail de Liu et al. 2018, qui sera publié en Mars 2018 dans la revue Cell : Rescue of Fragile X Syndrome Neurons by DNA Methylation Editing of the FMR1 Gene (Guérison des neurones syndrome de l’X fragile par édition via CRISPR de la méthylation de l’ADN sur le gène FMR1).
Edition épigénétique et génétique par CRISPR : le gène FMR1 et le syndrome de l’X fragile (Liu, Cell, Mars 2018)
Le système CRISPR-Cas9 a été initialement développé pour l’édition génomique chez les mammifères, Figure 2 (pour plus de détails, voir sur ce site : CRISPR-Cas9 édition génomique).
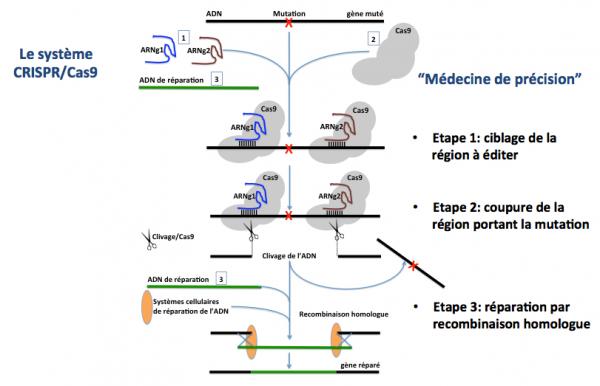
Figure 2 : CRISPR-Cas9 dans sa version classique.
Dans ce travail, le système CRISPR-Cas9 a été utilisé dans deux approches différentes afin de restaurer l’expression du gène FMR1 et de normaliser sa fonction physiologique dans des cellules humaines : i) par déméthylation des cytosines (C) de la séquence CGGn ainsi que celles des ilots CpG du promoteur du gène, ii) par raccourcissement de la séquence répétée de triplets CGG.
Déméthylation de la séquence CGGn et réactivation de FMR1 dans le syndrome de l’X fragile
Dans une première approche, les auteurs ont utilisé une lignée cellulaire (FX52 iPSC) issue de cellules d’un malade porteur du syndrome de l’X fragile. Dans ces cellules, le gène FMR1 contient une séquence répétée de 450 triplets CGG et son niveau d’expression est de 0,3% par rapport à celui observé dans des cellules humaines normales.
Ces cellules FX52 iPSC ont été infectées avec un lentivirus permettant l’expression de dCas9-TET1 et d’un ARN guide (ARNg) ciblant la séquence GGCGGCGGCGGCGGCGGCGGNGG.
La protéine recombinante dCas9-TET1 est une protéine hybride construite par génie génétique (Choudhurry 2016) issue de la fusion entre une protéine Cas9 inactivée (dCas9, d pour dead) et le domaine catalytique de TET1. TET1 est une enzyme de la famille des Ten-Eleven Translocation dioxygenases. Ces protéines sont impliquées dans la déméthylation de l’ADN en convertissant la 5-méthyl-Cytosine en 5-hydroxyméthylcytosine, puis finalement par d’autres étapes en Cytosine (Guo 2011).

Figure 3 : Déméthylation de la séquence des triplets CGG et réactivation de l'expression du gène FMR1. Dans cette approche, (1) un ARN guide (ARNg) et (2) une protéine hybride constituée d'une version inactivée de Cas9 (dCas9) et du domaine catalytique de la TET1 sont co-transfectés dans les cellules issues de malades atteints du syndrome de l'X fragile (CGGn avec n=450). L'ARNg guide la protéine dCas9-TET1 sur la séquence des triplets répétés de FMR1 et la TET1 en déméthyle les cytosines, ainsi que les cytosines des ilots CpG. L'expression du gène est alors activée.
Dans cette version, l’ARNg “guide” la protéine hybride dCas9-TET1 sur la séquence CGG de FMR1 et TET1 déméthyle les cytosines (Figure 3). Les résultats montrent que le taux de méthylation des C passe de 100% dans les cellules FX52 iPSC non traitées à seulement 4% dans les cellules traitées par CRISPR, et dans le même temps le niveau d’expression de l’ARNm FMR1 passe de 0,3 à 90% et la production de la protéine FMRP est rétablie. Des résultats identiques ont été obtenus avec d’autres lignées cellulaires humaines issues de malades atteints du syndrome de l’X fragile.
Les auteurs ont également montré que les effets “hors cible” de cette approche sont limités à 29 locus génétiques pour lesquels le niveau de changement est faible lorsqu’il existe.
Les cellules FX52 iPSC peuvent donner des neurones lorsqu’on les soumet à un protocole de différenciation spécifique (Chambers 2009). Le niveau d’expression de FMR1 dans les neurones exprimant le système CRISPR (dCas9-TET1 et ARNg) était de 82% de celui observé dans les neurones normaux ; cependant, FMR1 n’était pas exprimé dans les neurones non traités avec le système CRISPR ou ceux traités avec une version inactivée de TET1. Ces résultats suggèrent que la différenciation des cellules XF52 iPSC en neurones n’affecte en rien l’expression et la réactivation de FMR1. De plus, lorsque ces neurones étaient greffés dans le cerveau de souris, les chercheurs ont observé qu’une fraction de ces neurones continuait à exprimer FMR1, montrant en cela que la correction de la méthylation par CRISPR se maintenait sur le long terme.
Raccourcissement de la séquence CGGn et réactivation de FMR1 dans le syndrome de l’X fragile
Dans une deuxième approche le système CRISPR-Cas9 a été utilisé pour raccourcir la séquence répétée de triplets CGG afin de restaurer l’expression du gène FMR1 et de normaliser sa fonction physiologique. Pour cela les chercheurs ont utilisé des cellules FXS iPSC issues de cellules de malades atteints du syndrome de l’X fragile avec plus de 450 CGG dans le gène FMR1 (Xie 2016). Après traitement avec le système CRISPR-Cas9, la méthylation du promoteur du gène était réduite de 39% et l’expression de l’ARNm et la protéine FMRP était restaurée (Figure 4).
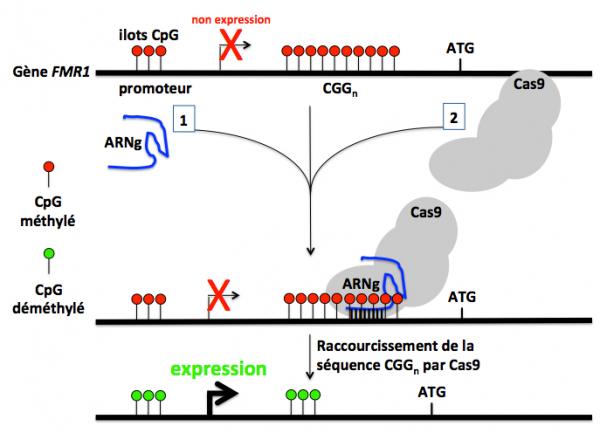
Figure 4 : Raccourcissement de la séquence des triplets CGGn et réactivation de l'expression du gène FMR1. Dans cette approche, (1) un ARN guide (ARNg) et (2) la protéine Cas9 sont co-transfectés dans les cellules issues de malades atteints du syndrome de l'X fragile (CGGn avec n=450). L'ARNg guide la protéine Cas9 sur la séquence des triplets répétés de FMR1. La Cas9 clive le gène et après réparation en résulte un raccourcissement qui au final est suivi d'une déméthylation des cytosines de la séquence répétée et des ilots CpG. L'expression du gène est alors activée.
Discussion
Notre connaissance des mécanismes par lesquels le syndrome de l’X fragile se met en place est encore insuffisante du fait de la complexité des circuits neuronaux et de leur implication dans le phénotype cérébral.
Par contre, on sait que lorsque la séquence répétée du triplet CGG comporte plus de 230 unités dans le gène FMR1, les cytosines (C) contenues dans cette séquence et dans les ilots CpG du promoteur du gène sont hyperméthylées avec pour conséquence une inhibition d’origine épigénétique de l’expression du gène. L’absence de la protéine FMRP est aujourd’hui considérée comme la cause principale du syndrome de l’X fragile. Cependant, le mécanisme par lequel la longueur anormale de la séquence CGG influe sur l’hyperméthylation des cytosines n’est pas élucidé.
Pour l’heure, il n’y a pas de traitement du syndrome de l’X fragile.
Les résultats présentés dans cet article fournissent la preuve directe que la déméthylation des C dans les séquences CGG répétés par le système CRISP est suffisante pour réactiver le gène FMR1 et la production de la protéine FMRP.
Pour aussi encourageants qu'ils soient, ces résultats ont été obtenus sur des lignées cellulaires, donc in vitro. Cette thérapie sera-t-elle aussi efficace lorsqu'elle aura été appliquée in vivo sur des modèles animaux ? Enfin, il reste à savoir si et quand ce type de thérapie sera utilisé pour le traitement de malades atteints de ce syndrome.
A suivre…
Bibliographie
Cliquer sur les noms des auteurs pour accéder à la page relative à l'article sur PubMed.
Les articles en accès libre sont noté d'un *.
Guo 2011 *
Xie 2016 *
Date de dernière mise à jour : 08/10/2018
Ajouter un commentaire