
Maladie de Huntington
Cette maladie est aussi appelée Chorée de Huntington.
La chorée est une manifestation d’atteinte neurologique se traduisant par des mouvements anormaux, incontrôlés et involonatires. Il s’agit d’une maladie monogénique transmise sur le mode autosomique dominant, mais dans 5 à 8% des cas, la maladie résulte d’une mutation de novo (qui n’était pas présente chez les parents).
Le gène responsable est HTT, situé sur le chromosome 4 (4p16.3). Il comprend 180 000 paires de bases et est constitué de 67 exons. Ce gène code pour la protéine Huntingtine (HTT). Dans sa forme normale, le gène comporte dans sa partie 5’ (“début” de la séquence codante) au niveau du premier exon, une séquence répétée du triplet CAG dont la longueur est comprise entre 16 et 25. Le triplet CAG code l’acide aminé glutamine dont l’abbréviation à une ou trois lettres est Q ou Gln. Le gène HTT code pour la protéine HTT qui comporte dans sa partie N-terminale (“début” de la séquence de la protéine) une séquence répétée de l’acide aminé glutamine -Q-Q-Q-Q-Q- (ou -Gln-Gln-Gln-Gln-Gln-). Cette séquence est dite “polyQ”.

Dans cette figure les acides aminés sont représentés par l'abbréviation à une lettre. Les différents exons dans le gène (en bas) et les parties correspondantes dans la protéine (en haut) sont représentés en noir et en bleu. Les séquences répétées polyQ et triplet CAG sont surlignées et soulignées en rouge. Dans cet exemple la longueur de la séquence polyQ est de 21 (individu “normal”).
La longueur de cette séquence polyQ est de 16 à 25 chez les individus “normaux”. La maladie n’apparaît que lorsque le nombre de glutamines excède 40. En fait, ce nombre de répétitions peut aller jusqu’à 120 chez certains malades. Généralement, plus la longueur de la séquence polyQ est importante et excède 40, plus l’âge d’apparition de la maladie diminue, et plus sa sévérité augmente.
Par exemple un père de 30 ans avec une séquence répétée de CAG de 45 unités reste encore asymptomatique pour une dizaine d’années. Cependant, si une expansion de cette séquence à 50-60 unités s'est produite lors de la méiose durant la spermatogénèse, son fils peut souffrir de la forme juvénile de la maladie avant son géniteur.
Les individus porteurs d’une séquence polyQ dont la longueur est comprise entre 27 et 35 semblent constituer un groupe particulier présentant les signes cliniques atténués de la maladie plus ou moins sévère selon les cas. Cependant, à côté de la HTT mutée (HTT-polyQ>40), il semble qu’il existe d’autres facteurs génétiques et/ou environnementaux favorisant ou au contraire atténuant la sévérité de la maladie comme le suggère le fait que dans une même famille et pour une séquence polyQ de même longueur, l’âge d’apparition des symptômes et la sévérité de la maladie sont variables.
La prévalence de cette maladie au niveau mondial est de l’ordre de 2,7 cas pour 100 000 personnes (Müller 2017), mais elle varie géographiquement fortement en fonction de la proportion d’individus Caucasiens. Il semblerait que cette maladie soit originaire de populations Nord Européennes (prévalence 10 à 14 pour 100 000). La prévalence est plus faible en Asie (1 à 7 par million). A l’inverse, la prévalence la plus élevée a été observée au Venezuela dans plusieurs villages de pêcheurs sur la côte Ouest du lac Maracaibo.
Deux versions différentes s’affrontent sur l’origine de la maladie au Venezuela.
- Dans la première version, toutes les familles touchées ont une ancêtre commune, Maria Concepcion Soto, qui vivait au début du 18eme siècle et dont le gène HTT muté aurait été hérité de son père, un navigateur Espagnol. Maria Conception mourut de la maladie et la transmit à 10 générations de descendants.
- Selon la seconde version, Antonio Doria, prêtre sur un navire Espagnol débarqua en 1867, dans le village de San Francisco et décida de s’y installer. Après avoir abandonné la prêtrise, il épousa une femme locale et ils eurent de nombreux enfants. En fait, Doria portait le gène HTT muté et il mourut de la maladie dix ans plus tard.
Aujourd’hui la prévalence de cette maladie dans la région de la côte Ouest du lac Maracaibo est la plus élevée au monde avec 1 cas sur moins de cent personnes (Wexler 2004)
Les autres régions de forte prévalence sont la Tasmanie (effet fondateur dans le contexte insulaire), l’Ecosse, le Pays de Galles et la Suède.
La maladie se manifeste le plus généralement entre 35 et 50 ans par trois types de troubles cliniques
- troubles moteurs avec présence d’une chorée, c’est à dire de mouvements involontaires, anormaux et incontrolables ; ces troubles progressent inexorablement avec la maladie.
- troubles cognitifs et troubles de la mémoire, contribuant fortement aux difficultés du malade et qui comme les troubles moteurs progressent avec la maladie.
- troubles comportementaux avec désordre émotionnel et changement de personnalité, associant des troubles du sommeil, des difficultés d’élocution et des difficultés à avaler. Ces troubles comportementaux ne sont pas toujours présents et ne semblent pas progresser avec la maladie.
La mort apparaît classiquement entre 15 et 20 ans après les premiers symptomes, et il faut signaler un taux élevé de suicides (10% des cas) notamment durant la période qui fait suite à la connaissance du diagnostic.
Le gène HTT est exprimé dans tous les tissus et en particulier dans le système nerveux central notamment au niveau du striatum, du cortex, de l’hippocampe et du cervelet, et est nécessaire au développement embryonnaire. La mutation responsable de la maladie (séquence polyQ d’une longueur supérieure à 40) n’entraîne pas un défaut d’expression de la HTT (comme c’est le cas par exemple dans la mutation F508Del sur la protéine CFTR qui est responsable de la mucoviscidose) mais plutôt la production d’une protéine aux fonctions anormales du fait de la mutation (séquence de polyQ>40).
La HTT “normale” est une longue protéine de 3144 aminoacides. Ses fonctions sont multiples et non encore totalement élucidées. Cette protéine intervient dans :
-
la dynamique cellulaire, c’est à dire dans l’architecture du cytosquelette, l’endocytose, le trafic intracellulaire des organelles (autophagosomes, endosomes, lysosomes) dans les axones et neurites, et l’adhésion cellulaire
-
le métabolisme cellulaire en particulier au niveau des mitochondries (production d’énergie dans les cellules) et la maladie diminue le transport du glucose dans les neurones notamment au niveau du striatum
-
la division cellulaire en interagissant avec le fuseau mitotique et les microtubules
-
l’homéostasie des protéines (autophagie)
-
l’expression de certains génes, notamment ceux codant pour des facteurs neuronaux comme le BDNF (brain derived neurotrophic factor) qui joue un rôle important dans la fonction des neurones et la neurogénèse.
Un aspect essentiel de la physiopathologie de cette protéine mutée est la formation de sous-fragments (exon 1) qui non seulement sont inactifs pour les fonctions attendues de la protéine mais aussi peuvent modifier le fonctionnement d’autres protéines. De plus, ces sous-fragments forment des aggrégats intracellulaires (pouvant contenir jusqu’à 100 000 unités) qui perturbent les fonctions des neurones et peuvent même entraîner leur mort par apoptose. En particulier, il semble que plus la longueur de la séquence polyQ est importante et plus la formation des aggrégats est efficace et rapide, et plus leur action est sévère. Cet aspect est réminiscent d’autres maladies neurodégénératives comme la maladie d’Alzheimer ou la maladie de Parkinson dans lesquelles des aggrégats protéiques semblent jouer un rôle patophysiologique important.
Toutes les fonctions de la HTT citées ci-dessus sont altérées par la présence de la séquence répétée de glutamine dans la protéine mutée (HTT-polyQ). Par exemple, la HTT normale stimule l’expression et le transport intraneuronal du BDNF nécessaire au fonctionnement des neurones au niveau des synapses, mais en présence de la protéine mutée cette stimulation est perdue. Ainsi, dans la maladie de Huntington, la HTT mutée a non seulement perdu sa capacité de stimuler la production et le transport du BDNF, mais aussi elle agit de façon dominante sur la protéine normale, inhibant sa fonction. Un autre exemple concerne les réactions d’excitotoxicité qui apparaissent consécutivement à la destruction neuronale. Les deséquilibres qui en résultent entre les neurotransmetteurs et leurs récepteurs au niveau des synapses conduisent à plusieurs réactions en chaîne : entrée massive de l’ion calcium dans les neurones et autres cellules neurales, activation d’enzymes (phospholipases, protéases, calpaïnes), dégradation des cellules et de leur contenu.
Ces exemples (et d’autres) montrent que la séquence répétée de polyQ a des conséquences négatives sur la fonction de la HTT normale et ses fonctions. De fait, un seul allèle muté suffit à causer la maladie. Il est intéressant ici de mentionner le syndrome de Wolf–Hirschhorn. Dans cette maladie les sujets atteints ont perdu une partie du chromosome 4p qui porte le gène HTT. Comme les malades Huntington, il n'ont que la moitié théorique du niveau de la HTT, mais ne présentent pas les symptomes de la maladie de Huntington (Harper 1996). Cet argument conforte le fait que la HTT mutée a un effet délétère sur la HTT normale.
On peut résumer la situation de la façon suivante : d’une part, les malades Huntington expriment un allèle muté et un allèle normal, de sorte que la protéine HTT normale n’est présente qu’à une concentration de 50% de son niveau théorique et ceci peut suffire au déclanchement de la maladie. D’autre part, la protéine HTT mutée a acquis des fonctions toxiques “nouvelles” qui se manifestent de façon dominante. De plus, la possibilité que la HTT mutée se transmette d’une cellule à une autre, de la même façon que le prion, a également été évoquée.
La présence de la Huntingtine mutée se traduit principalement par la perte importante de neurones dans le striatum à un stade avancé de la maladie. Le striatum est une région sous-corticale impliquée dans les mouvements volontaires, les motivations et recherche de plaisir (sexuel, alimentaire, etc.), et la neurogénèse (réparation et régénération neuronale). La mort neuronale caractérise pleinement la phase ultime de la maladie bien que les dysfonctionnements neuronaux qui donnent naissance aux signes cliniques majeurs, la prédèdent largement. Par exemple en terme de troubles moteurs, la chorée (mouvements involontaires et incontrôlables) semble traduire des dysfonctionnements neuronaux tandis que la bradykinésie (lenteur des mouvements volantaires) serait plutôt corrélée à la mort neuronale (Ross 2014).
L’examen des malades Huntington en IRM cérébrale montrent une importante perte de volume du striatum dans les phases précoce (bien avant l’apparition des signes cliniques) et finale de la maladie. Cette perte de volume est d’autant plus importante que la longueur de la séquence de polyQ est élevée dans la protéine HTT mutée, et elle corrèle avec les marqueurs cliniques des troubles moteurs et des fonctions cognitives mais pas avec ceux des troubles psychiatriques. Des pertes de volume sont aussi enregistrées au niveau d’autres aires du cerveau, pallidum, thalamus, hippocampe, et cortex, mais leurs conséquences sont moins dramatiques que celles qui affectent le striatum.
Le diagnostic de cette maladie par analyse génétique de la mutation (nombre des répétitions CAG > 40) sur le gène HTT peut être réalisé bien avant l’apparition des premiers symptômes. A l’heure actuelle il n’existe pas de traitement définitif de cette maladie, mais de nombreuses études sont en cours.
Nouvelles molécules en cours d’essai :
- sur les troubles moteurs : pridopidine, deutetrabenazine.
- sur le cours de la maladie : ISIS443139, rilmenidine et selisistat (diminution de l’expression de la HTT mutée), laquinimod (réduit l’apoptose et induit la régénération cellulaire), PBT2 (inhibe la formation des aggrégats de sous-fragments de la HTT mutée)
La thérapie cellulaire (transplantation de cellules neurales non porteuses de la mutation) au niveau du striatum semble prometteuse, mais les quelques essais réalisés chez l’homme n’ont pas donné de résultats suffisamment convaincants et des études expérimentales chez l’animal sont encore nécessaires.
Edition génomique: éliminer la forme mutée de la Huntingtine
Rappelons que dans une maladie génétique à transmission dite dominante, la présence d’un seul allèle muté suffit à déclancher la maladie. L’élimination pure et simple du produit de cet allèle muté (la huntingtine mutée) permettrait donc de guérir la maladie et le système CRISPR/Cas9 représente à cet égard une fantastique possibilité thérapeutique. C’est ce que vient de démontrer le groupe du Dr. Beverly Davidson du Children Hospital de Philadelphie (article publié en janvier 2017).
Les chercheurs ont imaginé une stratégie qui permet de n’éliminer que la forme mutée de la protéine Huntingtine (codée par l’allèle muté), tout en gardant présente sa forme normale (allèle normal). Pour cela ils ont utilisé le fait que le gène HTT muté (par expansion de la séquence CAG) comporte également d’autres mutations sur un seul nucléotide notamment dans sa région régulatrice (par exemple C->G). Il s’agit de ce qu’on appelle un “polymorphisme d’un seul nucléotide” (en anglais “single nucleotide polymorphism” ou SNP). De telles mutations (SNPs) sont extrêmement fréquentes, plusieurs millions dans le génome humain. Elles peuvent avoir des conséquences fonctionnelles importantes si elles modifient le cadre de lecture ou la nature de l’acide aminé du codon. Ces SNPs constituent la base de la susceptibilité à certaines maladies.
Nous avons déjà vu que le système CRISPR/Cas9 nécessite deux critères : premièrement la complémentarité d’hybridation entre la séquence ciblée sur l’ADN génomique et la séquence de l’ARN guide, et deuxièmement la présence d’un motif PAM au voisinage de la région d’hybridation avec l’ADN génomique ciblé (en position 3’). Ce motif PAM est essentiel pour le positionnement correct de la Cas9 et son action ultérieure. Et c’est sur ces motifs PAM que se sont concentrés les chercheurs : si un SNP crée un site PAM sur l’allèle muté, l’allèle normal (non porteur du PAM) sera insensible au système CRISPR/Cas9. La séquence PAM de Cas9 est : 5’-NRG-3’, avec N représantant tout nucleotide, R un nucléotide purine (c’est à dire A ou G), et G la guanine. Exemples de séquences PAM/Cas9: AAG, AGG, TAG, TGG, CGG, GGG, etc. Tous ces triplets représentent donc des sites potentiels de reconnaissance par Cas9.
Berverly Davidson et ses collaborateurs ont identifié divers SNPs dans le gène HTT en association préférentielle (en déséquilibre de liaison) avec l’expansion CAG>35. Certains de ces SNPs sont présents chez une proportion importante des malades. Par exemple, dans la région régulatrice (promoteur) du gène HTT normal, il existe une séquence GGC. Elle ne sera pas reconnue comme un motif PAM par la Cas9. Par contre, dans le gène HTT muté qui comporte l’expansion CAG >35, cette séquence GGC est mutée en GGG (c’est un SNP). On peut alors utiliser cette séquence PAM GGG pour ne cibler que l’allèle muté du gène HTT et laisser inaffecté l’allèle normal. Pour cela, les chercheurs ont synthétisé des ARN guides qui ciblent spécifiquement une séquence proche du PAM dans la région du gène HTT muté en position 5’ de l’exon 1 et dans l’intron 1. En effet, la séquence polyQ (poly glutamine) issue des répétitions CAG se trouve au tout début de la protéine (dans sa partie N-terminale) et cette séquence est responsable en partie de la toxicité de la huntingtine mutée. Ainsi comme le montre la figure ci-dessous et suivant cette stratégie, le système CRISPR/Cas9 permet d’éliminer l’exon 1 dans l’allèle du gène HTT muté, évitant ainsi la production de la protéine mutée, sans affecter l’allèle normal, la protéine HTT normale continuant à être produite.
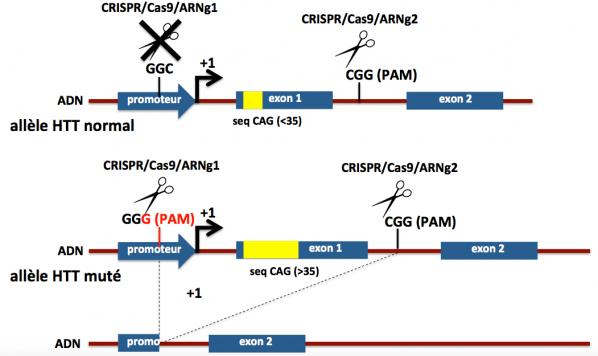
Stratégie CRISPR/Cas9 pour la maladie de Huntington. Adapté de Monteys 2017
Ne sont représentés sur cette figure que les deux premiers exons du gène HTT (1 et 2) qui en comporte en réalité 67. La flèche à angle droit notée +1 matérialise le début de la transcription du gène HTT. Les introns sont représentés en marron et les exons en bleu. La séquence de répétitions CAG est figurée en jaune. On voit qu’après action du système CRISPR/Cas9 l’exon 1 et une partie du promoteur de l’allèle muté sont éliminés. La HTT tronquée produite (manque la partie codée par l’exon 1) est peu ou pas produite (promoteur tronqué également) et vraisemblablement dégradée. Noter que la cassure générée au niveau de l’intron 1 (entre les exons 1 et 2) de l’allèle normal est réparée après action du système CRISPR/Cas9 et il n’y a pas de conséquence sur la production de la protéine HTT normale.
Pour démontrer l’efficacité de cette stratégie, les auteurs ont utilisé un modèle de souris transgénique (BacHD) dans laquelle le gène humain HTT muté (contenant les SNPs ciblés dans cette stratégie) est exprimé. Ces souris présentent tous les symptômes de la maladie humaine. Les souris ont été traitées par un virus adéno associé recombinant exprimant la protéine Cas9 et les ARNg1 et g2, injecté dans l’hémisphère droit du cerveau. Pour vérifier l’effet de la thérapie, l’hémisphère gauche n’a pas reçu le traitement et était utilisé comme contrôle de l’expérience. Les cerveaux des souris ont été prélevés 3 semaines plus tard et l’ADN a été isolé et analysé pour la présence ou l’absence de la cassure du gène HTT. De même l’ARN a été isolé et analysé pour la présence de l’ARN messager codant pour la HTT. La figure ci-dessous montre les résultats de cette étude.
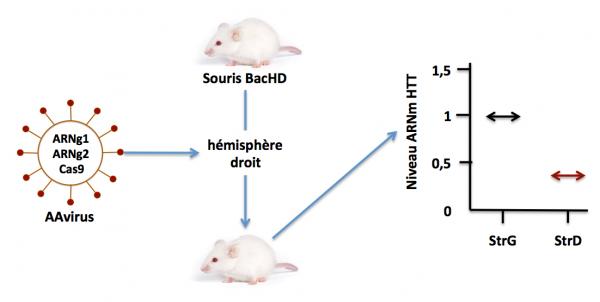
Stratégie CRISPR/Cas9 pour la maladie de Huntington. Confirmation in vivo. Adapté de Monteys 2017
On voit que l’ARNm HTT dans le striatum (StrD) de l’hémisphère droit est réduit de plus de 60% par rapport à son niveau dans le striatum (StrG) de l’hémisphère gauche (contrôle non injecté). Le fait que le niveau d’ARNm HTT ne soit pas nul dans le striatum droit montre que le système CRISPR/Cas9 n’a pas fonctionné à 100% et que 40% des allèles mutés n’ont pas été “édités” dans certaines cellules du striatum. Il reste donc beaucoup à faire.
De plus, et en absence d’une étude plus complète sur les paramètres et marqueurs biologiques de la maladie chez les animaux traités, il n’est pas possible de savoir si ce traitement a entraîné une guérison partielle ou non de la maladie.
Dans un article plus récent (Yang et al. 2017) le groupe de Xiao-Jiang Li, Emory University School of Medicine à Atlanta, montre que l’élimination de la HTT mutée dans le cerveau d’un modèle de souris par le système CRISPR/Cas9 ciblant spécifiquement l'exon 1 de HTT dans les neurones (et non les autres types cellulaires cérébraux) permet de corriger divers paramètres biologiques de ces souris (performances motrices, force, masse corporelle) suggérant ainsi que la thérapie CRISPR/Cas9 est efficace sur cette maladie.
Bibliographie
Date de dernière mise à jour : 20/09/2023
Ajouter un commentaire