
CRISPR/Cas9 édition génomique
Un outil basé sur le système immunitaire adaptatif des bactéries et archées
Proies de toutes sortes d’agressions biologiques, incluant les phages, virus, plasmides, et autres congénères, les bactéries et archées ont mis en place au cours de leur évolution des systèmes de défense leur permettant de détecter, reconnaître et éliminer des ADN étrangers différents de leur propre ADN afin de survivre dans un environnement potentiellement hostile.
Les systèmes CRISPR (Clustered Regularly Interspaced Small Palindromic Repeats ; traduction approximative en Français : Courtes répétitions palindromiques groupées et régulièrement espacées) font partie de cet arsenal en tant que systèmes immunitaires adaptatifs. Ils sont présents dans le génome de 90% des archées et de plus de 40% des bactéries. Les systèmes CRISPR sont absents du génome des eukaryotes et des génomes viraux.
Première mise en évidence
En 1987, des chercheurs Japonais (Ishino et al.) découvrent des séquences répétées dans le gène iap de la bactérie Echerichia coli
En séquençant le gène Iap (phosphatase alcaline) de la bactérie Echerichia coli, ces chercheurs ont mis en évidence la présence de séquences répétées dans la région 3’-non codante du gène. Ces séquences étaient séparées par d’autres séquences qui s’avèreront plus tard être des séquences d’ADN de phages (virus infectant les bactéries) et/ou de plasmides, vestiges d’infections anciennes. Ces résultats restèrent inexpliqués pendant de nombreuses années.
Cependant, il devint rapidement évident que de telles séquences répétées existaient dans de nombreux microorganismes (bactéries et archées).
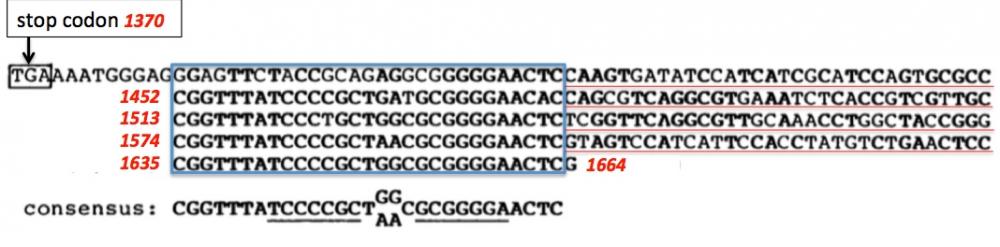
Séquences répétées dans le gène Iap
Figure 1. Seule la partie 3’-non codante du gène Iap qui se termine par le nucléotide G en position 1664, est représentée ici. Le codon stop (TGA, position 1370) marque la fin de la traduction de l’ARN messager Iap en protéine. La suite de la séquence n’est pas traduite en protéine mais généralement elle joue un rôle important par exemple dans la stabilité du messager. Ici, ce qui a attiré l’attention des chercheurs est la découverte de séquences répétées en position 1379, 1452, 1513, 1574 et 1635 (encadrées en bleu). On notera qu’entre ces séquences répétées on trouve d’autres séquences (soulignées en rouge). La séquence “consensus” des séquences répétées est soulignée en noir : TCCCCGCT--CGCGGGGA. On peut voir que les deux demi-séquences soulignées forment un palindrome. Les séquences répétées sont les “small palindromic repeats” et les autres séquences sont les “spacers” dans le jargon CRISPR (Clustered Regularly Interspaced Small Palyndromic Repeats). Les “spacers” sont dérivés de séquences génomiques de phages ou de plasmides. Adapté de Ishino 1987.
Ces résultats restèrent inexpliqués pendant de nombreuses années. Cependant, il devint rapidement évident que de telles séquences répétées existaient dans de nombreux microorganismes (bactéries et archées).
Ce n’est qu’au début des années 2000 que le terme CRISPR a été utilisé pour désigner ces séquences répétées (Mojica 2000 ; Jansen 2002).
Exemple : le système CRISPR/Cas de Streptococcus thermophilus
Les figures ci-dessous montrent 3 des 4 systèmes CRISPR/Cas de la bactérie Streptococcus thermophilus, utilisée dans l’industrie agroalimentaire (fabrication de yogourts).
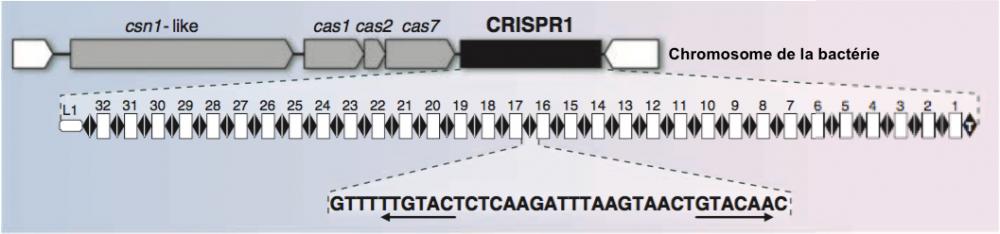
Système CRISPR1 de la bactérie S. thermophilus
Figure 2. Partie haute : vue partielle du chromosome de la bactérie. Le locus CRISPR est représenté par un rectangle noir. Les gènes associés à CRISPR, csn1-like, cas1, cas2 et cas7, sont représentés en gris, ils codent pour les protéines Cas (CRISPR associated).
Partie centrale : le locus CRISPR est délimité à ses deux extrêmités par la séquence leader (L1) et par l’élément terminal (T). Les “repeats” sont figurés par des couples de lozange noirs. Les “spacers” sont figurés par des rectangles blancs numérotés de 1 à 32.
Partie basse : la séquence des “repeats” avec (soulignée par les flêches) les deux éléments du palindrome. Adapté de Horvath 2010.
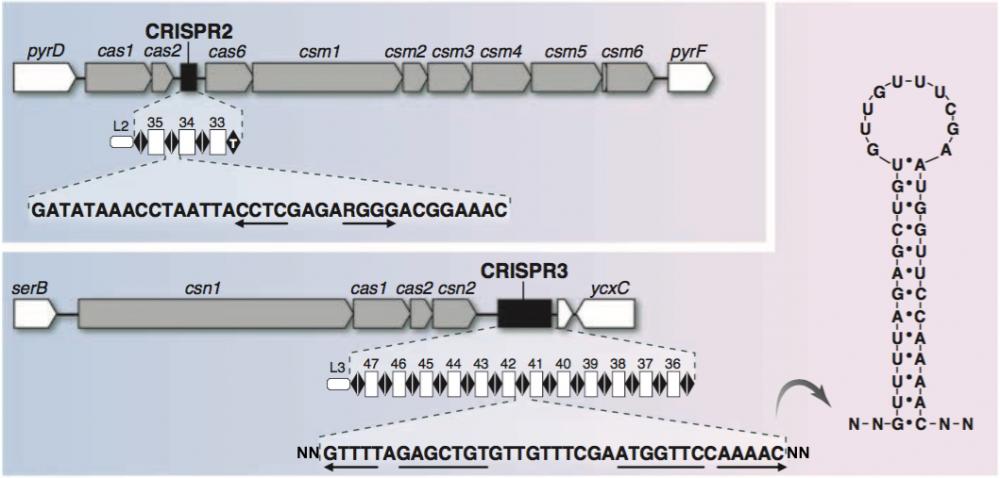
Systèmes CRISPR2 et 3 de la bactérie S. thermophilus
Figure 3. Le locus CRISPR2 et les gènes cas et csm associés sont encadrés par les gènes bactériens pyrD et pyrF. Le locus CRISPR3 et les gènes cas et csm associés sont encadrés par les gènes bactériens serB et ycxC. La partie basse de la représentation de CRISPR3 montre la séquence des “repeats” dans laquelle les éléments palindromiques sont soulignés et flêchés.
A droite : la struture palindromique formée par les “repeats”. Noter l’appariement des bases A-U et C-G. Adapté de Horvath 2010.
Comment ça marche chez les microorganismes ?
Nous allons voir ci-dessous comment le système CRISPR/Cas fonctionne dans les bactéries et archées.
Le mécanisme CRISPR/Cas comporte deux étapes majeures : immunisation et aquisition de l’immunité.
Etape d’immunisation (figure ci-dessous)
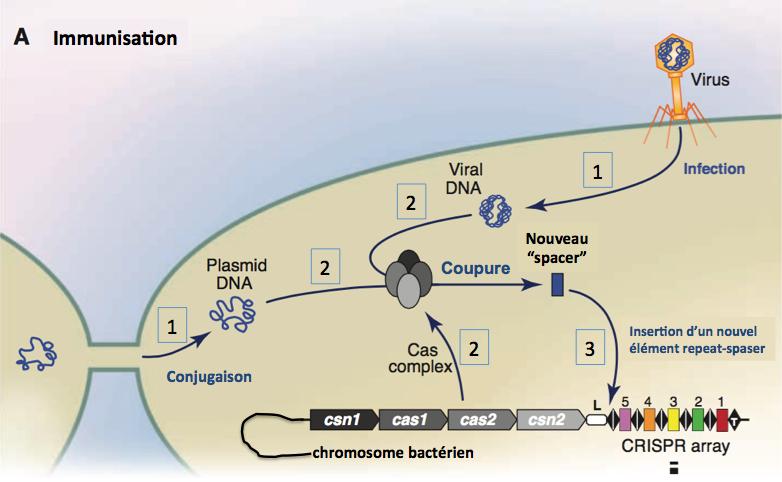
Adapté de Horvath 2010.
Figure 4.
1. L’ADN viral (après infection par un phage) ou plasmidique (après conjugaison avec une autre bactérie) est libéré dans la bactérie hôte.
2. Cet évènement conduit à l’expression des protéines Cas qui interagissent et forment un complexe. Ce complexe lie l’ADN étranger/invasif au voisinage d’une séquence PAM (proto-spacer adjacent motif, voir plus loin) et le clive en fragments (spacers). On ne sait pas comment ces deux étapes (expression des Cas et clivage) sont régulées, mais les protéines Cas1 et Cas2 sont clairement impliquées.
3. Le nouveau “spacer” produit est inserré dans le locus CRISPR sous forme d’un élément repeat-spacer en aval de la séquence leader (L). Ainsi le locus CRISPR s’allonge, infection après infection. Il apparaît donc que le spacer 1 résulte d’une infection plus ancienne que celle ayant donné naissance au spacer 2 et ainsi de suite. L’analyse des divers éléments “repeat-spacer” permet donc d’évaluer la chronologie des épisodes d’infection de la bactérie.
Etape d’acquisition de l’immunité (figure ci-dessous)
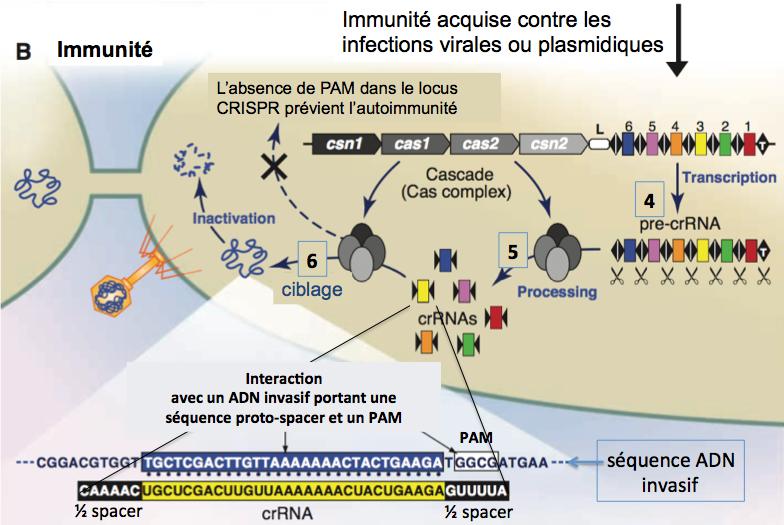
Adapté de Horvath 2010.
Figure 5.
4. L’ADN CRISPR est transcrit en ARN sous le contrôle de la séquence leader (L) qui agit comme promoteur.
5. Le pre-CRISPR RNA (pre-crRNA) est clivé en éléments “partial repeat-spacer-partial repeat”, les crRNAs, par un complexe multiprotéique de protéines Cas.
6. Les crRNAs liés sur le complexe multi-Cas activent la machinerie CRISPR/Cas en s’hybridant par leur séquence “spacer” à l’ADN cible dont ils ont été dérivés lors d’une précédente infection (voir la partie basse de la figure). Le complexe Cas dégrade l’ADN (ou ARN) invasif, conduisant à son inactivation, éliminant ainsi l’agent infectieux.
A noter que les crRNAs ciblent aussi bien les brins sens et anti-sens de l’ADN invasif. L’analyse des séquences de phage adjacentes aux proto-spacers (qui vont donner les spacers) révèle la présence en 3’ de séquences conservées appelées CRISPR motifs ou proto-spacer adjacent motifs (ou PAM). Pour qu’une séquence de phage soit ciblée par CRISPR, il faut donc qu’elle soit au voisinage d’un motif PAM (voir partie basse de la figure ci-dessus dans laquelle ce motif est GGCG). Cependant, ce motif PAM ne fait pas partie du spacer, il n’est pas présent dans le locus CRISPR. Ceci permettrait d’éviter l’action du système CRISPR sur le génome de l’hôte.
En résumé
Le système CRISPR se caractérise de la façon suivante
- Les combinaisons des CRISPR et des gènes Cas forment les systèmes CRISPR/Cas.
- Présence de multiples séquences “repeats” de 23 à 47 paires de bases, identiques avec peu ou pas de variation. Ces séquences forment le plus souvent des palindromes (haute stabilité). Leur nombre est généralement inférieur à 50.
- Présence de “spacers” de même taille mais de séquences très différentes entre les demi-“repeats” (longueur de l’ordre de 21 à 72 paires de bases). Ces séquences présentent un haut degré de similarité avec des séquences trouvées dans le génome de phages ou dans des plasmides.
- Chaque “spacer” est encadré par 2 demi-“repeats”.
- Présence de séquences “leader” de quelques centaines de paires de bases et d’un “terminal repeat” qui encadrent le locus CRISPR.
- Présence de gènes divers Cas, Cas1 à Cas4, codant pour des protéines portant des fonctions typiques de nucléase, hélicase, polymérase et de liaison aux polynucléotides.
- Les locus CRISPR sont le plus souvent retrouvés sur le chromosome (plus rarement sur les plasmides).
- Plusieurs locus CRISPR peuvent être trouvés dans un même microorganisme (bactérie ou archée).
- La fonction majeure du système CRISPR/Cas des microorganismes est de détruite le génome d’agents invasifs (phages, virus, plasmides).
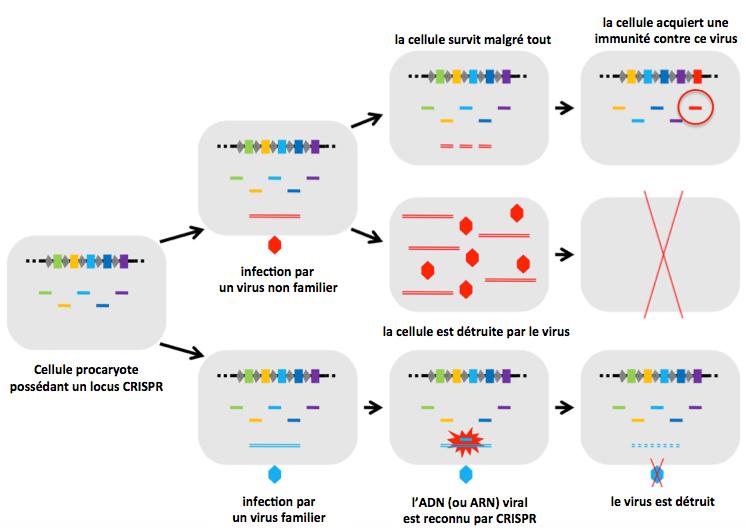
Figure 6. Impact du système CRISPR/Cas sur la survie des procaryotes (bactéries ou archées) en réponse aux virus (phages).
Lors de l’infection par un virus (familier, hexagone bleu) déjà rencontré par la bactérie, le système CRISPR/Cas reconnaît et détruit l’intrus. La bactérie survit. Dans le cas d’une première infection par un virus (non familier, hexagone rouge), la bactérie meurt ou s’adapte en acquérant un nouveau spacer (entouré de rouge). Dès lors, elle pourra répondre efficacement à une nouvelle infection par ce virus grâce à son système CRISPR/Cas en utilisant le spacer adapté (rouge sur la figure).
Adapté de Koonin 2009.
Vers les applications de cette technique
Le système CRISPR/Cas9 de type II, exemple de Streptococcus pyogenes
Il existe trois types différents de systèmes CRISPR-Cas, les types I, II et III, qui utilisent des mécanismes moléculaires différents pour cibler l’ADN invasif et le dégrader. Le motif PAM, courte séquence adjacente à la séquence cible de l’ADN invasif, joue un rôle décisif dans les systèmes CRISPR de types I et II. Les systèmes de types I et III utilisent des complexes regroupant diverses protéines Cas (voir l’exemple du système de S termophilus dans les figures précédentes) pour la dégradation de l’ADN invasif. En revanche, le type II requiert une seule protéine Cas (Cas9) pour la reconnaissance de l’ADN invasif et sa dégradation, une caractéristique qui s’est révélée extrêmement utile pour les applications sur l’édition du génome, comme nous allons le voir plus loin.
La bactérie Streptococcus pyogenes utilise un système CRISPR/Cas9 de type II
En 2011 Delcheva et al. rapportent qu’un petit ARN codé par un gène du locus CRISPR en amont du gène Cas9 est essentiel pour la maturation du pre-crARN (voir figure ci-dessous). En fait, cet ARN appelé transactivating crARN (tracrARN) se lie à Cas9 et s’hybride aux “repeats” du pre-crRNA. Ces hybrides tracrARN:pre-crARN sont clivés par la ribonucléase III (RNase III, figure) pour générer des hybrides tracrARN:crARN. Voir la figure ci-dessous.
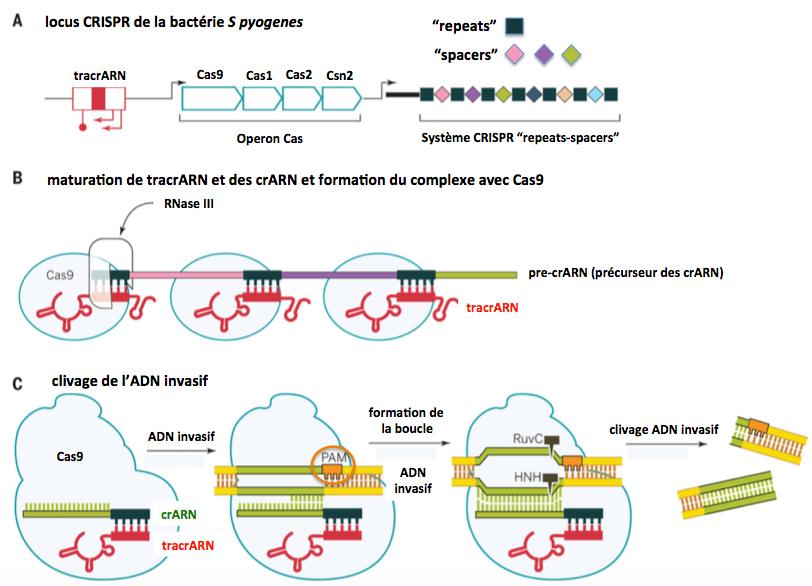 Figure 7.
Figure 7.
Adapté de Doudna 2014
Les hybrides tracrARN:crARN générés restent fixés sur la Cas9, où se fait le recrutement de l’ADN invasif par hybridation avec la séquence “spacer” du crARN. La protéine Cas9 possède deux domaines catalytiques HNH et Ruv-C qui, arès ouverture du double brin de l’ADN du génome invasif au niveau de la région complémentaire au crARN (formation de la boucle), coupent respectivement le brin complémentaire et le brin codant de l’ADN invasif, générant ainsi une coupure double brin. Cette coupure se fait trois nucléotides en amont (ou en aval selon le brin) de la séquence PAM. Pour le système CRISPR/Cas9 de S. pyrogenes cette séquence PAM est NGG* sur le brin ciblé (CCN* sur le brin complémentaire), et la séquence ciblée par le crARN est GN20GG.
Les systèmes CRISPR différents utilisent des PAM différents: S. pyrogenes: NGG*; S.thermophilus NGGNG et NNAGAAW*; S. mutans NGG ou NAAR*.
*N: tout nucleotide; W: A ou T; R: A ou G.
La protéine Cas9 est donc “guidée” dans son action par deux ARN, le tracrARN et le crARN adapté à l’ADN invasif ciblé. L’équipe d’Emmanuelle Charpentier et Jennifer Doudna a imaginé et construit un ARN chimérique unique, tracrARN-crARN, appelé ARN guide en liant ces deux ARN par une courte séquence (linker loop sur la figure ci-dessous). Cet ARN guide qui peut être synthétisé en laboratoire permet de cibler tout ADN en adaptant la séquence du crARN (Jinek 2012). Ce point est très important pour les applications de CRISPR/Cas9 à l'édition du génome.
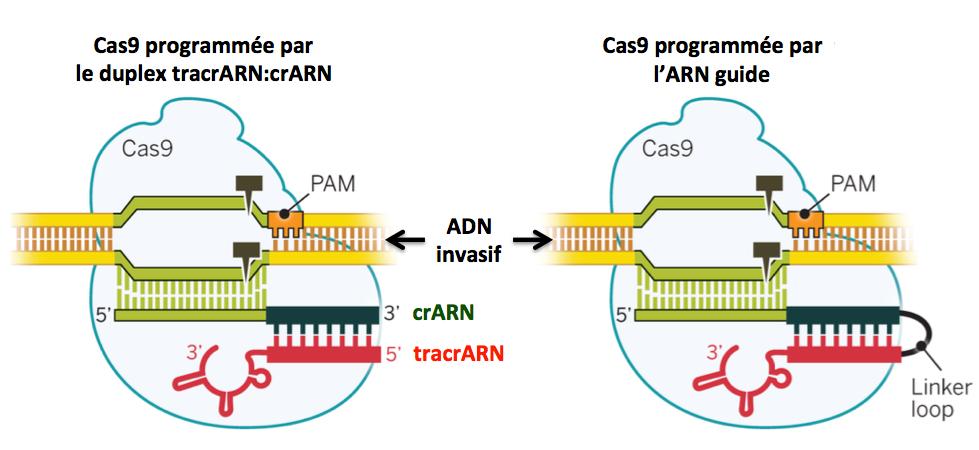
Figure 8.
Adapté de Doudna 2014
En résumé,
L'utilisation de CRISPR/Cas9 met en jeu un ARN guide ciblant la protéine Cas9 sur un ADN génomique dont la séquence est complémentaire de celle de la partie crARN du guide. La Cas9 coupe alors l'ADN à la position souhaitée, définie par la séquence de la partie crARN du guide. C'est la base de l'édition génomique.
Quelles utilisations peuvent être faites de CRISPR/Cas9 ?
A la suite des travaux d’Emmanuelle Charpentier et Jennifer Doudna, plusieurs groupes confirmèrent la pertinence de la technique décrite ci-dessus. En effet selon celle-ci, le système CRISPR/Cas9 peut être utilisé pour générer des coupures double brin dans l’ADN et pour cela il ne nécessite que deux entités, la protéine Cas9 et l’ARN guide (ARNg). Ces deux entités peuvent être facilement transfectées dans tout organisme (cellule ou in vivo) par utilisation de méthodes appropriées.
Le système CRISPR/Cas9 induit des cassures double brin de l’ADN et c’est là le point essentiel de son utilisation pour l’édition génomique.
Réparation des cassures double brin
Les cassures qui affectent simultanément les deux brins d’ADN qui forment la double hélice, comme celles résultant par exemple de l’action de radiations ionisantes (rayons X, rayons gamma, etc.) sont les lésions les plus léthales que les cellules aient à subir. Si ces cassures ne sont pas réparées, elles peuvent conduire à des réarrangements genomiques/chromosomiques qui seront le point de départ d’un processus de transformation (cancer) ou de mort cellulaire.
Lorsqu’une cassure double brin apparaît sur l’ADN, les systèmes cellulaires de réparation interviennent rapidement et vont opérer suivant deux mécanismes différents :
- recombinaison homologue (HR, homologous recombination) par échange d’information génétique entre deux séquences identiques d’ADN,
- jonction d'extrémités non-homologue (NHEJ, non-homologous end joining), ce dernier processus générant des insertions ou délétions dans l’ADN “réparé”.
Ces deux processus sont décrits en détail dans les figures ci-dessous.
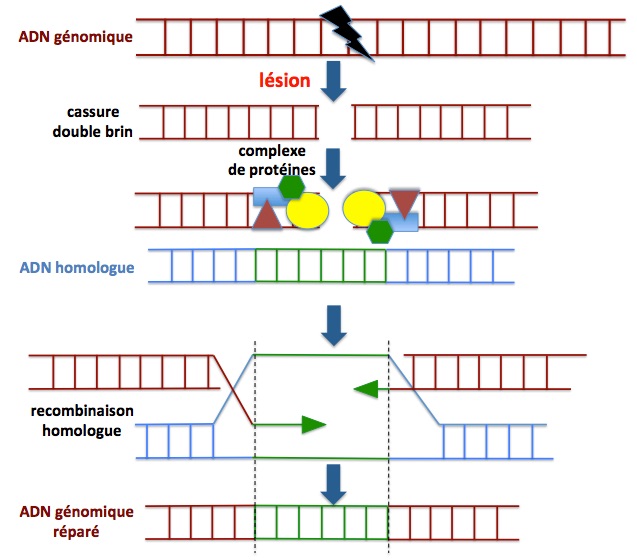
Figure 9. Réparation par recombinaison homologue. Ce mécanisme permet une réparation correcte de l’ADN par utilisation d’une chromatide sœur (fragment d’ADN dont la séquence est la même que celle de la partie de l’ADN lésé, les deux fragments étant dits “homolgues”) comme patron (matrice de réparation). Comme indiqué dans la partie basse de la figure, les deux brins du fragment d’ADN sont synthétisés par appariement avec les nucléotides de la séquence “patron” de l’ADN homologue. Cependant, ce mécanisme est restraint aux phases S et G2 du cycle cellulaire durant lesquelles une copie de chaque chromosome (dont un fragment constituera l’ADN homologue) est disponible. Comme on le verra plus loin, c'est la raison pour laquelle une séquence homologue (matrice de réparation) est apportée en même temps que l'ARNg et la Cas9 pour l'édition génique corrective. Un complexe de différentes protéines (Rad51, Mre11, Rad50, Nbs1, RPA, Rad52, BRCA1 et BRCA2) est impliqué dans ce processus de réparation à l’issue duquel la séquence de l’ADN lésé a été correctement corrigée. Adapté de Nicolai 2015.
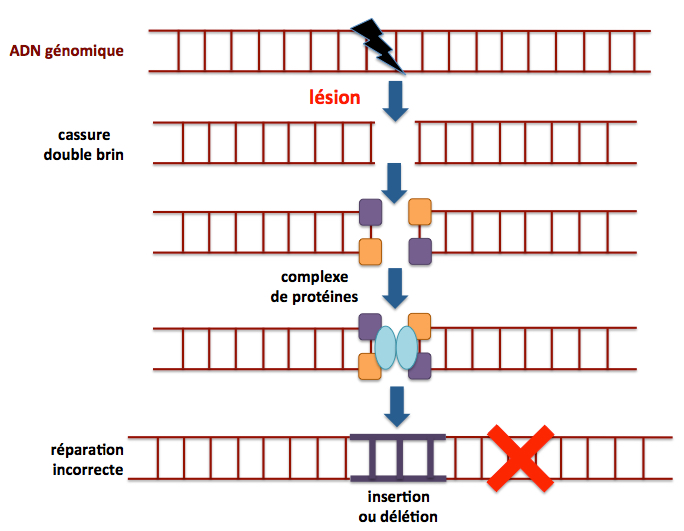
Figure 10. Réparation par jonction d'extrémités non-homologue. Ce mécanisme, à l’inverse de celui décrit précédemment, prédomine durant les phases G0 et G1 du cycle cellulaire, même s’il peut également intervenir dans les autres phases du cycle. Il est donc le mécanisme le plus utilisé pour la réparation de l’ADN chez les eucaryotes. Il consiste dans le simple raboutage des deux brins sans utilisation de patron. Il en résulte souvent une délétion (perte d’une partie de l’ADN à réparer) ou une insertion (addition d’une séquence autre que celle de l’ADN à réparer) qui constituent une source d’erreurs, contribuant à l’accumulation de mutations notamment au cours du vieillissement. Un complexe de différentes protéines (kinases ciblant l’ADN, l’hétérodimère Ku70-Ku86, la nucléase Artemis, les polymerases μ et λ, l’ADN ligase IV, XRCC4, XLF et PAXX4/XLS/c9orf142) est impliqué dans ce processus de réparation à l’issue duquel le gène concerné est le plus souvent rendu inactif (croix rouge) du fait de la présence d’une délétion ou d’une séquence incorrecte. Adapté de Nicolai 2015.
Il apparaît donc qu’au site de la cassure, la séquence initiale peut être modifiée ou une nouvelle séquence peut être inserrée. C'est sur ces deux points qu'est basée l'édition génomique.
Edition génomique chez les eucaryotes
Le système CRISPR/Cas9 n’est pas présent dans le génome des eucaryotes. Si on souhaite modifier le génome d’un eucaryote par ce système il est donc nécessaire de l'y amener c'est à dire d’apporter l’ARN guide (spécifique de la région ciblée), la protéine Cas9 et la matrice de réparation (ADN homologue) selon le but recherché. Le groupe de George Church a été un des pionniers dans ce domaine (Mali 2013, Mali 2014).
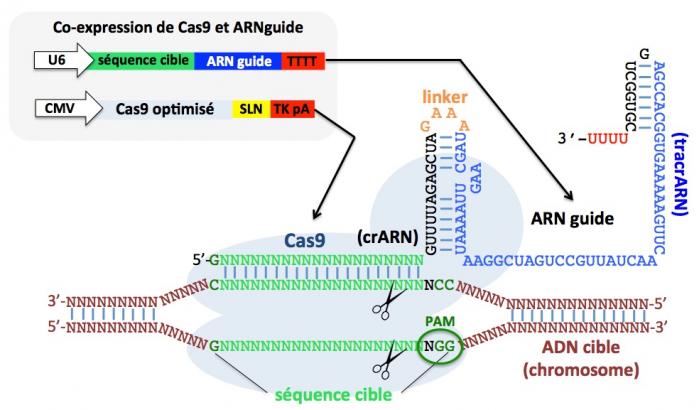
Figure 11. Edition génomique dans des cellules humaines par utilisation du système CRISPR/Cas9
Cette méthode est basée sur la co-expression de deux constructions, l’une produisant l’ARN guide (ciblant la séquence d’ADN à modifier en vert sur la figure) sous le contrôle du promoteur U6 de la polymérase III humaine, l’autre produisant la protéine Cas9 (en bleu clair sur la figure) optimisée pour les séquences humaines (et contenant une séquence de localisation nucléaire, SLN) sous le contrôle du promoter CMV (cytomegalovirus). Toute séquence (en vert) telle que GN20GG (N, tout nucléotide) sera ciblée à condition qu’un motif PAM (cerclé en vert foncé) soit présent en position 3’. TkpA: signal de polyadenylation de la thymidine kinase. On peut voir que l’ARN guide est composé du crARN (qui cible la séquence à modifier, partie en vert et en noir) lié au tracrARN (bleu) par le linker (orange). A noter que plusieurs ARN guides (ciblant des séquences d’ADN différentes) peuvent être utilisés simultanément. Adapté de Mali et al. 2013.
Les applications du système CRISPR-Cas9
Les applications du système CRISPR/Cas9 sont multiples et du fait que cette technologie permet de toucher au génome humain, certaines de ses applications sont extrêmement prometteuses, mais d’autres en revanche posent de sérieux problèmes au plan éthique. Ce sont ces deux aspects “Dr Jekyll” et “Mr Hyde” que nous allons voir maintenant.
CRISPR/Cas9 : “Le côté Dr Jekyll”
Il existe des milliers de maladies rares (voir Maladies Rares sur ce site), le plus souvent incurables et d’origine génétique dues à une ou plusieurs mutations dans un ou plusieurs gènes. Le seul espoir de guérison de telles maladies est la thérapie génique, c’est à dire la correction de la ou des mutations responsables. La technologie CRISPR/Cas9 qui permet la modification du génome et notamment la correction de mutations génétiques, représente donc un immense espoir dans ce domaine.
Les applications les plus directes de l’édition génomique pour le grand public sont celles qui consistent à traiter ces maladies comme par exemple la mucoviscidose, la dystrophie de Duchenne ou la maladie de Huntington, pour ne citer que celles-ci.
Maladies monogéniques recessives : correction par recombinaison homologue
Ces maladies, telles que la mucoviscidose, la dystrophie musculaire de Duchenne ou la drépanocytose, résultent d’une ou plusieurs mutations dans un gène dont la conséquence est la dégradation de la protéine codée par ce gène ou son inactivation (voir sur ce site Maladies Rares, “Les diverses Causes”). L’utilisation du système CRISPR/Cas9 permet de corriger l’allèle muté en remplaçant la séquence comprenant la mutation par une séquence identique ne comprenant pas la mutation. L’exemple présenté sur la figure ci-dessous pourrait s’appliquer au cas de la mutation F508Del dans le gène CFTR, responsable de 80% des cas de mucoviscidose.
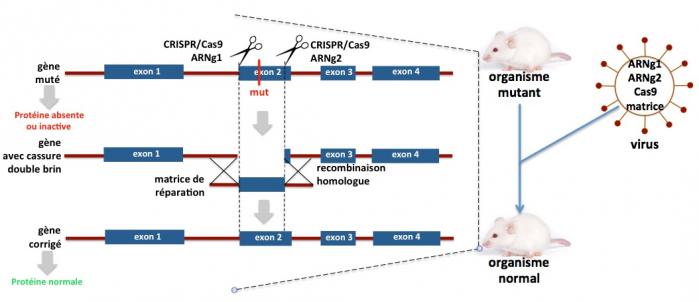
Figure 12. Dans cet exemple, un gène comportant 4 exons est le siège d’une mutation dans l’exon 2 (barre verticale rouge, mut) qui rend la protéine soit instable (absente), soit inactive. Les systèmes CRISPR/Cas9 ARNg1 (ciblant le début de l’exon2) et CRISPR/Cas9 ARNg2 (ciblant la partie droite de l’exon 2) produisent une délétion de la majeur partie de l’exon 2 contenant la mutation. En présence de la matrice de réparation de séquence identique à la partie délétée de l’exon 2 mais ne contenant pas la mutation, la recombinaison homologue permet de corriger le gène muté. La protéine est alors exprimée normalement et active. Pour réaliser cette correction, les ARN guides g1 et g2, la protéine Cas9 et la matrice de réparation doivent être apportés dans l’organisme par un vecteur, le plus souvent un virus adéno-associé recombinant (partie de la figure à droite). Ce type de virus à ADN est non pathogène pour l’homme.
Maladies monogéniques recessives : correction par jonction d’extrémités non homologues
Exemple de la correction de la dystrophie musculaire de Duchenne (DMD). La DMD est due à des mutations dans le gène DMD (chromosome Xp21.2) qui conduisent à un déficit complet en dystrophine, une protéine sub-sarcolémique des muscles squelettiques. Le gène DMD est un des plus longs du génome humain (2,3 millions de paires de nucléotides). Il comporte 79 exons. Sa transcription en prémessager prend 16 heures. La dystrophine est une protéine de 3685 acides aminés. Ces mutations sont le plus souvent responsables d’un décalage du cadre de lecture de l’ARN correspondant, du fait de la délétion d’un ou de plusieurs exons ou de mutations dans un exon.
Un exemple de décalage du cadre de lecture est présenté ci-dessous. Chacun des acides aminés qui constituent la séquence de la protéine est codé par trois nucléotides dans la séquence du gène et de l’ARN correspondant (noter que dans l’ARN, l’uracyle U remplace la thymidine T). CUC code pour la leucine, GUA pour la valine, et ainsi de suite (voir le dossier Maladies Rares, page : les notions de base). La mutation est figurée par une barre oblique dans la séquence du gène. Elle provoque un décalage de la phase de lecture, ce qui entraîne la production d’une protéine de séquence différente et donc inactive, dégradée ou absente.
CTC GTA TTT AGC CGT séquence du gène
CUC GUA UUU AGC CGU séquence de l’ARN
Leu Val Phe Ser Arg Protéine normale
C/TCG TAT TTA GCC GT séquence du gène
C UCG UAU UUA GCC GU séquence de l’ARN
Ser Tyr Leu Ala Protéine de séquence différente
(inactive ou dégradée)
CT/CGT ATT TAG CCG T séquence du gène
CU CGU AUU UAG CCG U séquence de l’ARN
Arg Ile Stop Arrêt de la traduction du fait du codon
TGA, un des codons stop (protéine
tronquée ou absente)
Plusieurs groupes de recherche ont montré qu’il était possible de corriger ce type de mutation par jonction d’extrémités non homologues.
Bien que l’intégrité de la séquence de toute protéine soit nécessaire à son fonctionnement normal, la perte d’une petite partie de sa séquence (et en particulier dans le cas d’une protéine aussi longue que la dystrophine) peut n’affecter que modérément son fonctionnement. C’est la stratégie envisagée dans le traitement de la DMD. Les exemples présentés ci-dessous montrent trois modes de correction par CRISPR/Cas9 (Ousterout 2014).
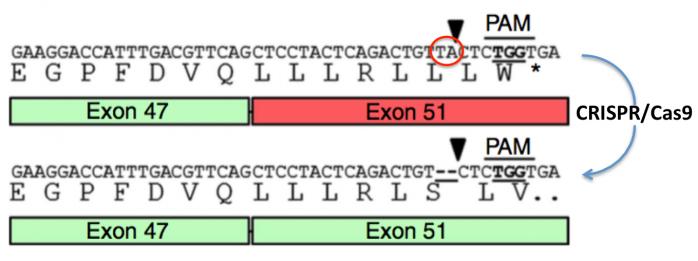
Figure 13. Etudes réalisées sur des myoblastes (cellules musculaires) d’un malade (Ousterout 2014).
Chez ce malade, il existe une délétion des exons 48, 49 et 50 qui n’aurait entraîné à elle seule qu’une forme atténuée de la maladie. En revanche, une mutation (addition de deux nucléotides T et A; cercle rouge) dans l’exon 51 produit un codon Stop prématuré (TGA) entraînant la formation d’une dystrophine tronquée inactive et rapidement détruite (donc absente des muscles).
Séquence mutée: ---CTC AGA CTG TTA CTC TGG TGA---
Leu Arg Leu Leu Leu Trp Stop*
Séquence normale: ---CTC AGA CTG TCT CTG GTG A---
Leu Arg Leu Ser Leu Val
On peut voir (partie haute de la figure) que l’introduction de deux nucléotides TA à gauche de la pointe de flèche (cercle rouge) provoque un décalage du cadre de lecture qui produit un codon stop (TGA) trois codons plus loin (*). Dans cette approche, les chercheurs ont utilisé le motif PAM (TGG) pour cibler cette mutation et l’éliminer, rétablissant ainsi le cadre de lecture et la séquence normale de la protéine. Bien qu’il manque les exons 48-50, ce malade ne présenterait qu’une forme atténuée de la maladie après cette correction.
Une autre stratégie possible consiste à éliminer complètement l’exon porteur de la mutation, dans l’exmple ci-dessous l’exon 51, ou les exons porteurs de mutations.
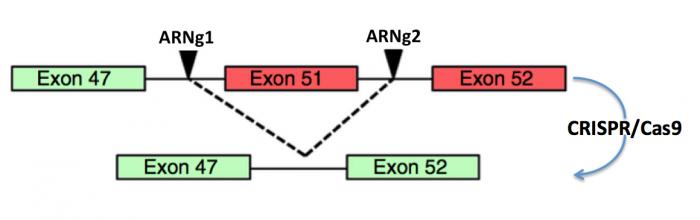
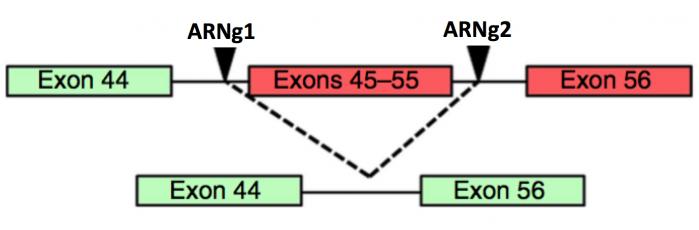
Figure 14. Dans ces cas, deux ARN guide (ARNg1 et ARNg2) sont synthétisés de telle sorte qu’ils encadrent l’exon (51) ou les exons (45-55) porteurs de mutations. A noter ici que ces délétions entraînent un raccourcissement de la protéine qui n’affecte que modérément son activité et le malade corrigé de cette façon ne présenterait qu’une version atténuée de la dystrophie.
Maladies monogéniques dominantes : le cas de la maladie de Huntington. Eliminer la forme mutée.
La maladie de Huntington est une maladie neurodégénérative fatale à transmission dominante. Elle résulte d’une expansion de répétion du triplet de nucléotides CAG dans l’exon 1 du gène HTT qui code pour la huntingtine (voir sur ce site Maladie de Huntington). Bien que la huntingtine soit exprimée dans toutes les cellules du corps, c’est la neuropathologie résultante de sa forme mutée qui est fatale. Elle est caractérisée par une atrophie progressive et irréversible du striatum, suivie par une perte plus ou moins importante du volume d’autres régions du cerveau. Des études chez l’animal ont montré que l’élimination de la forme mutée représente une stratégie thérapeutique de choix pour les malades.
Rappelons que dans une maladie génétique à transmission dite dominante, la présence d’un seul allèle muté suffit à déclancher la maladie. L’élimination pure et simple du produit de cet allèle muté (la huntingtine mutée) permettrait donc de guérir la maladie et le système CRISPR/Cas9 représente à cet égard une fantastique possibilité thérapeutique. C’est ce que vient de démontrer le groupe du Dr. Beverly Davidson du Children Hospital de Philadelphie (article publié en janvier 2017).
Les chercheurs ont imaginé une stratégie qui permet de n’éliminer que la forme mutée de la protéine Huntingtine (codée par l’allèle muté), tout en gardant présente sa forme normale (allèle normal). Pour cela ils ont utilisé le fait que le gène HTT muté (par expansion de la séquence CAG) comporte également d’autres mutations sur un seul nucléotide notamment dans sa région régulatrice (par exemple C->G). Il s’agit de ce qu’on appelle un “polymorphisme d’un seul nucléotide” (en anglais “single nucleotide polymorphism” ou SNP). De telles mutations (SNPs) sont extrêmement fréquentes, plusieurs millions dans le génome humain. Elles peuvent avoir des conséquences fonctionnelles importantes si elles modifient le cadre de lecture ou la nature de l’acide aminé du codon. Ces SNPs constituent la base de la susceptibilité à certaines maladies.
Nous avons déjà vu que le système CRISPR/Cas9 nécessite deux éléments : premièrement la complémentarité d’hybridation entre la séquence ciblée sur l’ADN génomique et la séquence de l’ARN guide, et deuxièmement la présence d’un motif PAM au voisinage de la région d’hybridation avec l’ADN génomique ciblé (en position 3’). Ce motif PAM est essentiel pour le positionnement correct de la Cas9 et son action ultérieure. Et c’est sur ces motifs PAM que se sont concentrés les chercheurs : si un SNP crée un site PAM sur l’allèle muté, l’allèle normal (non porteur du PAM) sera insensible au système CRISPR/Cas9. La séquence PAM de Cas9 est : 5’-NRG-3’, avec N représantant tout nucleotide, R un nucléotide purine (c’est à dire A ou G), et G la guanine. Exemples de séquences PAM/Cas9: AAG, AGG, TAG, TGG, CGG, GGG, etc. Tous ces triplets représentent donc des sites potentiels de reconnaissance par Cas9.
Berverly Davidson et ses collaborateurs ont identifié divers SNPs dans le gène HTT en association préférentielle (en déséquilibre de liaison) avec l’expansion CAG>35. Certains de ces SNPs sont présents chez une proportion importante des malades. Par exemple, dans la région régulatrice (promoteur) du gène HTT normal, il existe une séquence GGC. Elle ne sera pas reconnue comme un motif PAM par la Cas9. Par contre, dans le gène HTT muté qui comporte l’expansion CAG >35, cette séquence GGC est mutée en GGG (c’est un SNP). On peut alors utiliser cette séquence PAM GGG pour ne cibler que l’allèle muté du gène HTT et laisser inaffecté l’allèle normal. Pour cela, les chercheurs ont designé des ARN guides qui ciblent spécifiquement une séquence proche du PAM dans la région du gène HTT muté en position 5’ de l’exon 1 et dans l’intron 1. En effet, la séquence polyQ (poly glutamine) issue des répétitions CAG se trouve au tout début de la protéine (dans sa partie N-terminale) et cette séquence est responsable en partie de la toxicité de la huntingtine mutée. Ainsi comme le montre la figure ci-dessous et suivant cette stratégie, le système CRISPR/Cas9 permet d’éliminer l’exon 1 dans l’allèle du gène HTT muté, évitant ainsi la production de la protéine mutée, sans affecter l’allèle normal, la protéine HTT normale continuant à être produite.
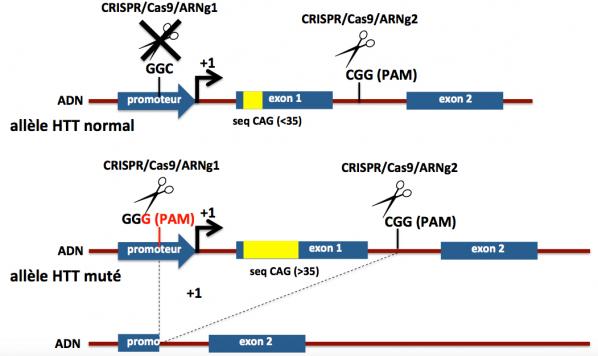
Figure 15. Adapté de Monteys 2017
Ne sont représentés sur cette figure que les deux premiers exons du gène HTT (1 et 2) qui en comporte en réalité 67. La flèche à angle droit notée +1 matérialise le début de la transcription du gène HTT. Les introns sont représentés en marron et les exons en bleu. La séquence de répétitions CAG est figurée en jaune. On voit qu’après action du système CRISPR/Cas9 l’exon 1 et une partie du promoteur de l’allèle muté sont éliminés. La HTT tronquée produite (manque la partie codée par l’exon 1) est peu ou pas produite (promoteur tronqué également) et vraisemblablement dégradée. Noter que la cassure générée au niveau de l’intron 1 (entre les exons 1 et 2) de l’allèle normal est réparée après action du système CRISPR/Cas9 et il n’y a pas de conséquence sur la production de la protéine HTT normale.
Pour démontrer l’efficacité de cette stratégie, les auteurs ont utilisé un modèle de souris transgénique (BacHD) dans laquelle le gène humain HTT muté (contenant les SNPs ciblés dans cette stratégie) est exprimé. Ces souris présentent tous les symptômes de la maladie humaine. Les souris ont été traitées par un virus adéno associé recombinant exprimant la protéine Cas9 et les ARNg1 et g2, injecté dans l’hémisphère droit du cerveau. Pour vérifier l’effet de la thérapie, l’hémisphère gauche n’a pas reçu le traitement et était utilisé comme contrôle de l’expérience. Les cerveaux des souris ont été prélevés 3 semaines plus tard et l’ADN a été isolé et analysé pour la présence ou l’absence de la cassure du gène HTT. De même l’ARN a été isolé et analysé pour la présence de l’ARN messager codant pour la HTT. La figure ci-dessous montre les résultats de cette étude.
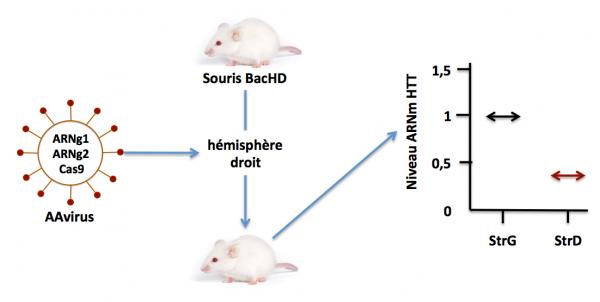
Figure 16.
On voit que l’ARNm HTT dans le striatum (StrD) de l’hémisphère droit est réduit de plus de 60% par rapport à son niveau dans le striatum (StrG) de l’hémisphère gauche (contrôle non injecté). Le fait que le niveau d’ARNm HTT ne soit pas nul dans le striatum droit montre que le système CRISPR/Cas9 n’a pas fonctionné à 100% et que 40% des allèles mutés n’ont pas été “édités” dans certaines cellules du striatum. Il reste donc beaucoup à faire.
De plus, et en absence d’une étude plus complète sur les paramètres et marqueurs biologiques de la maladie chez les animaux traités, il n’est pas possible de savoir si ce traitement a entraîné une guérison partielle ou non de la maladie.
Dans un article plus récent (publié en juillet 2017) le groupe de Xiao-Jiang Li, Emory University School of Medicine à Atlanta, montre que l’élimination de la HTT mutée dans le cerveau d’un modèle de souris par le système CRISPR/Cas9 ciblant spécifiquement l'exon 1 de HTT dans les neurones (et non les autres types cellulaires cérébraux) permet de corriger divers paramètres biologiques de ces souris (performances motrices, force, masse corporelle) suggérant ainsi que la thérapie CRISPR/Cas9 est efficace sur cette maladie.
CRISPR/Cas9 : “Le côté M. Hyde”
Modification du génome d'embryons humains
C'est l'utilisation du système CRISPR/Cas pour modifier le génome d'un embryon humain qui pose un problème éthique majeur.
La motivation d'une telle thérapie peut évidemment être tout à fait louable, comme par exemple la possibilité pour un couple (dont les deux membres sont porteurs de mutations dans un gène responsable d'une maladie incurable) d'avoir un enfant en bonne santé. Et c'est sur ce type de problème que seront réalisées les premières expérimentations dans les dix prochaines années.
Mais le risque de voir cette technologie dévoyée pour tenter notamment de créer des humains aux caractères nouveaux est bien présent. L'encadrement très strict de ces expérimentations par les agences nationales de régulation comme la FDA aux USA et l'EMA en Europe peut représenter un premier “garde fou” (Voir sur ce site: Nouvelles: La FDA tance John Zhang). Mais qu'en est-il de pays dans lesquels les aspects éthiques ne sont pas prioritaires ?
La Chine à cet égard est en première ligne.
Deux publications d’équipes Chinoises, parues en 2015 (Liang et al.) et 2016 (Kang et al.) rapportaient les premières expérimentations réalisées sur des zygotes humains (ovule fécondé, une seule cellule) surnuméraires, porteurs de 3 pronoyaux et donc non viables. Les résultats sur la correction de mutations dans le gène de la β-globine (HBB) qui code pour une sous-unité de l’hémoglobine et dont les mutations sont responsables des thalassémies, et sur l’insertion d’un allèle muté déficient du gène CCR5Δ32 le récepteur du HIV, montraient une efficacité médiocre avec un taux de correction d’environ 10% ou moins et un mosaïcisme des embryons, c’est à dire la présence de cellules embryonnaires ayant subi l’édition génomique et d’autres (cellules sauvages) ne l’ayant pas subie.
En Mars 2017 paraissait la publication d’un autre groupe Chinois rapportant l’utilisation de la méthode CRISPR/Cas9 sur des zygotes humains normaux. Dans ce travail (Tang et al.) les chercheurs ont utilisé dix zygotes spécialement générés pour l’étude à partir d’ovules de donneuses et de sperme d’un homme porteur d’une β-thalassémie (hétérozygote pour la mutation β41–42 dans le gène HBB) et dix zygotes à partir d’ovules de donneuses et de sperme d’un homme porteur d’une mutation G1376T dans le gène G6PD porté par le chromosome X. Tous les zygotes ont reçu une injection intracytoplasmique de la protéine Cas9 purifiée, d’un ARN guide ciblant la mutation et d’un ADN de correction (90 nucléotides) destiné à la correction de la mutation par recombinaison homologue. Les embryons ont été cultivés pendant deux jours avant analyse de leur génome. Les résultats ont montré que pour la correction de la mutation du gène G6PD, les deux embryons générés et porteurs de la mutation étaient tous deux corrigés (100%) par recombinaison homologue. Cependant l’un des deux comportait des cellules corrigées et des cellules non corrigées (mosaïque). Pour la mutation sur le gène HBB, 4 embryons sur 10 portaient la mutation et le taux de réussite de la correction par recombinaison homologue était de 50%. Enfin, l’analyse intégrale de tout le génome d’un des embryons porteur de la mutation sur G6PD montrait une absence de modification génomique autre que celle ciblée (G6PD).
Les auteurs concluaient : « Cependant, l’utilisation de CRISPR/Cas9 en thérapie génétique germinale n’est pas une option à l’heure actuelle du fait des problèmes techniques et éthiques qu’il reste à surmonter (aspects sécuritaire, mosaïcisme, etc.) ».
Cet article marque une étape notable vers les applications de la méthode à la thérapie génique germinale humaine.
En Août 2017 paraissait un quatrième article par le groupe de Soukrat Mitalipov (USA).
Leur travail a porté sur plus de cent embryons humains. Voir l'analyse de ce travail sur ce site dans Nouvelles: CRISPR/Cas et embryons humains.
A suivre...
Date de dernière mise à jour : 16/10/2017
Ajouter un commentaire