
L'enfant aux trois parents
ou
La thérapie par remplacement mitochondrial
On connaît plus de 7000 maladies rares dont 80% sont d’origine génétique. Dans l’immense majorité des cas, les gènes incriminés se trouvent dans le noyau des cellules sur les chromosomes. Mais certaines de ces maladies, très invalidantes et parfois mortelles, sont dues à des mutations affectant des gènes présents sur l’ADN contenu dans les mitochondries (Mitalipov 2014).
C’est de ces maladies et de leur thérapie que nous allons parler ici.
Un diaporama peut être téléchargé ICI.
Les mitochondries, des organelles venues de la nuit des temps…
Les mitochondries sont des organelles présentes dans les cellules eucaryotes, levures, champigons, plantes, animaux. Elles assurent, entre autres fonctions, la production d’énergie dans nos cellules.
Les mitochondries sont les vestiges d’une infection endosymbiotique d’une archée bactérie (ou d’un proto-eucaryote) par une a-protobactérie aérobique, survenue il y a plus d’un milliard d’années. De ce fait, les mitochondries ont gardé des ressemblances avec les bactéries (taille, forme, ADN circulaire, reproduction par fission, ribosomes 70S, chaîne transporteuse d’électrons, etc.). Elles possèdent leur propre génome, un ADN mitochondrial (ADNmt) circulaire d’environ 16500 paires de bases. Chaque mitochondrie abrite entre 2 et 10 copies d’ADNmt et on peut trouver jusqu’à 1000 mitochondries par cellules, en particulier dans les organes dont le fonctionnement requiert une grande quantité d’énergie, notamment les cellules cardiaques, musculaires et les neurones.
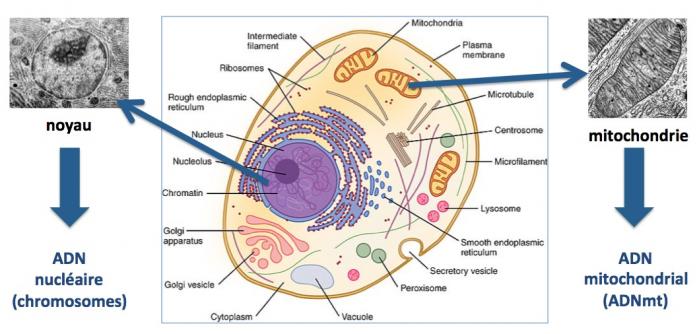
Bien que possédant leur propre génome, les mitochondries sont constituées d’environ 1200 protéines différentes qui dans leur immense majorité (98%) sont codées par des gènes nucléaires. Seulement 13 des protéines présentes dans les mitochondries sont codées par l’ADN mitochondrial. Mais ces protéines jouent un rôle majeur dans la production de l’ATP, le carburant cellulaire. Outre cette fonction vitale et essentielle, les mitochondries jouent également un rôle important dans d’autres fonctions comme l’apoptose (mort cellulaire programmée), l’homéostasie du calcium, la synthèse des hémes (hémoglobine, myoglobine, cytochromes) et des hormones stéroïdes. (Margulis 2006; Vafai 2012; Koonin 2015; O'Malley 2015; Gustafsson 2016).
Les mitochondries produisent l’ATP dont toutes nos cellules ont besoin
Dans leur grande majorité, les fonctions cellulaires ont besoin d’énergie comme par exemple la synthèse de l’ADN, de l’ARN, des protéines, des membranes cellulaires, la réparation de l’ADN, les mouvements cellulaires, le transport de molécules dans les cellules ou hors des cellules, etc.
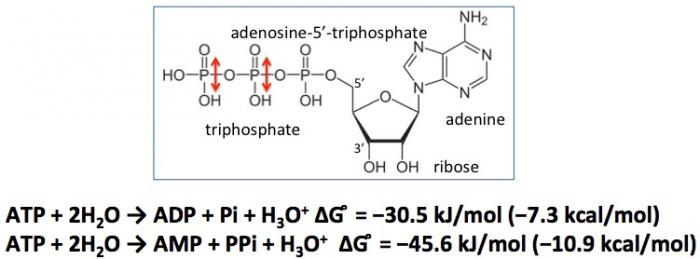
Cette énergie est apportée par l’adénosine triphosphate (ATP) lors de cassure des deux liaisons entre les groupes phosphate de la molécule (voir ci-dessus). Par ailleurs, l’ATP tient un rôle essentiel dans la signalisation cellulaire qui contrôle notamment l’expression des gènes.
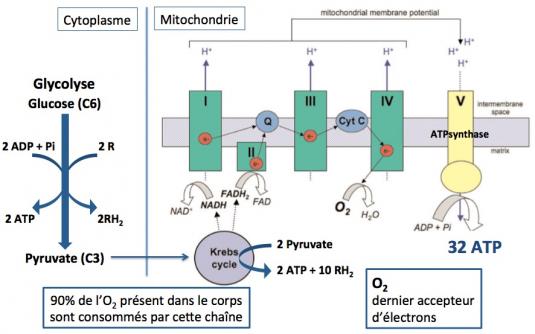
La synthèse de l’ATP nécessite une série de réactions de phosphorylation oxydative. La première étape est la glycolyse, c’est à dire la transformation du glucose en pyruvate qui a lieu dans le cytoplasme des cellules. Cette réaction fournit 2 molécules d’ATP et 2 molécules d’agent réducteur (RH2, NADH et FADH2). Le pyruvate entre dans la matrice de la mitochondrie et est pris en charge dans le cycle de Krebs (ou cycle de l’acide citrique) qui va générer 2 molécules d’ATP et 10 molécules d’agent réducteur (RH2). Ce sont ces agents réducteurs NADH et FADH2 qui vont alimenter la chaîne de phosphorylation oxydative en fournissant les électrons dont elle a besoin pour fonctionner, jusqu’à la production d’ATP par l’ATP synthase. NADH fournit ses électrons au complexe I et FADH2 au complexe II. Les électrons sont transportés vers le complexe III par le co-enzyme Q (Q) et vers le complexe IV (cytochrome C oxydase) par le cytochrome C. Au stade final l’oxygène est réduit en eau et 90% de l’oxygène de notre corps est utilisé pour la production d’ATP.
(Protti 2006)
A chaque étape de ces réactions un proton (H+) est transféré dans l’espace intermembranaire de la mitochondrie et un gradient de protons se crée. C’est ce gradient de protons qui va activer l’ATP synthase (complexe V) pour produire un total de 32 molécules d’ATP à partir de la phosphorylation de l’ADP. Les complexes I à V sont constitués de 85 protéines, dont 13 sont codées par l’ADNmt, 7 dans le complexe I, 1 dans le III, 3 dans le IV, et 2 dans le V. Toutes les protéines du complexe II sont codées par des gènes nucéaires.
Les mitochondries sont transmises par la mère
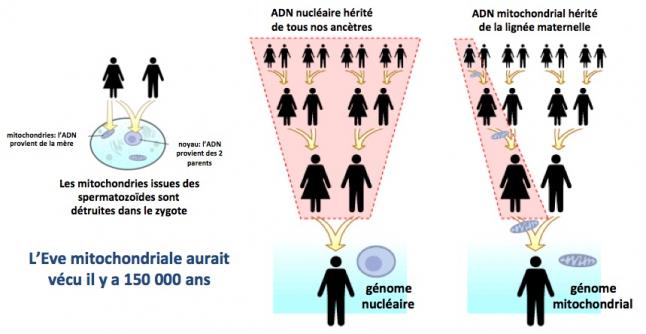
Contrairement à l’ADN nucléaire (chromosomes) qui est le résultat d’une combinaison entre l’ADN du père et l’ADN de la mère, L’ADN mitochondrial est transmis uniquement par la mère. Chez Caenorhabditis elegans (un minuscule ver très utilisé par les chercheurs comme eucaryote modèle), dès la fécondation de l’ovocyte, les mitochondries paternelles sont éliminées par autophagie, un processus complexe qui consiste à dégrader et éliminer toute structure biologique qui pourrait affecter la survie des cellules. Bien que ce processus n’ait jamais été démontré chez l’homme, l’absence de mitochondrie d’origine paternelle dans les cellules humaines suggére que ce processus est très ancien et a été conservé au cours de l’évolution. Ainsi, une mère portant une mutation dans son ADNmt peut la transmettre à toute sa descendance, filles et garçons, et seules les filles pourront à leur tour la transmettre à leur descendance. En revanche, les descendants de ses fils ne seront pas atteints par la maladie. (Wallace 2015)
Les mutations de l’ADNmt
Comme l’ADN nucléaire, l’ADNmt est sujet à de multiples agressions par des agents internes (espèces réactives de l’oxygène, erreurs de transcription, etc.) et externes (radiations ionisantes, carcinogènes, etc.). Ces agressions se traduisent par des mutations ou délétions qui affectent d’une part les ARN mitochondriaux de transfert (codés par l’ADNmt) et d’autre part les protéines mitochondriales de la chaîne de transport d’électrons impliquée dans la production d’ATP. Ces mutations sur l’ADNmt sont plus fréquentes que les mutations affectant l’ADN nucléaire car elles ne sont pas aussi efficacement réparées. Elles peuvent se produire dans la lignée germinale primitive, dans les ovogonies (cellules souches de ovocytes) ou dans les ovocytes et transmises de façon héréditaire à la descendance. Elles peuvent également se produire dans des cellules somatiques (muscle, système nerveux central, etc.) durant le développement et s’accumuler avec l’âge (une des causes du vieillissement). Plus de 250 mutations ou délétions ont été identifiées comme causes de maladies mitochondriales, et on estime qu’une personne saine sur 200 porte dans son ADNmt une des 10 mutations les plus fréquemment associées à ces maladies. (Mandavilli 2002; Taylor 2005; Saneto 2013; Lightowlers 2015)
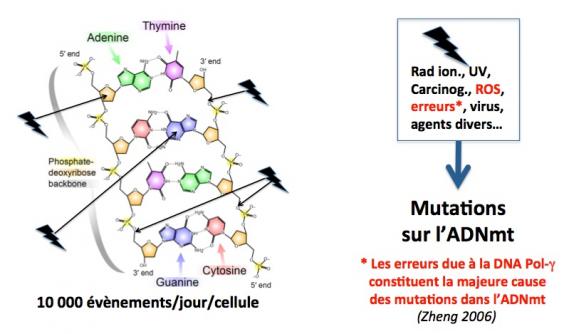
Homoplasmie et hétéroplasmie
Le génome mitochondrial est présent en une multitude de copies (plusieurs milliers) par cellules. De fait, deux situations sont possibles : homoplasmie ou hétéroplasmie. L’homoplasmie se réfère à la situation dans laquelle il n’y a pas d’accumulation significative de mutations. La séquence de toutes les copies de l’ADNmt est la même quelle que soit la nature du tissu ou des cellules. C’est le cas en général. A l’inverse, l’hétéroplasmie se manifeste par la présence dans les cellules de deux ou plusieurs copies différentes (ADNmt mutants) de l’ADNmt. Dans le cas d’une hétéroplasmie (et si l’ADNmt mutant est pathogène), il existe un niveau seuil au dessus duquel la maladie va apparaître. Ce seuil peut varier d’un malade à l’autre et d’un tissu à l’autre, d’un mutant à un autre, mais il est en général de l’ordre de 60-70% (c’est à dire que l’ADNmt mutant représente 60-70% de l’ADNmt total). En dessous de ce seuil, il est possible que l’individu, bien que porteur, ne soit pas malade.
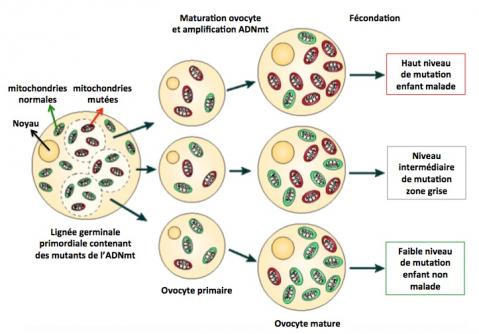
La notion d’homoplasmie est cependant plus apparente que réelle. En effet, il serait naïf de penser que tous les génomes mitochondriaux d’un organisme sont identiques. En fait, l’ADNmt est sans cesse l’objet de mutations qui sont transmises par expension mitochondriale, ou éliminées par macro-autophagie des mitochondries. Le fait qu’on ne détecte pas de mutants de l’ADNmt dans un prélèvement ne signifie pas qu’ils ne soient pas présents dans d’autres tissus ou d’autres cellules mais peut-être simplement qu’on ne les a pas trouvés car en trop faible nombre. (Taylor 2005; Stewart 2015; Picard 2016)
Les maladies mitochondriales
Les maladies mitochondriales touchent 1 naissance sur 4300 en Europe et aux USA. Elles affectent souvent plusieurs organes et en particulier les tissus dont le fonctionnement nécessite un fort apport énergétique comme le cerveau, les muscles, le cœur, le foie, etc. Elles sont généralement très hétérogènes associant surdité, cécité, faiblesse musculaire, insuffisance cardiaque, rénale, hépatique, autres. Comme dit précédemment, leur présentation clinique dépend du rapport ADNmt (muté)/ADNmt (normal) et ce rapport peut varier d’un tissu à un autre, ou d’un enfant à l’autre issu de la même mère.
Etant donné les fonctions importantes que jouent les mitochondries dans les cellules et les tissus, on peut comprendre que toute atteinte à leur fonctionnement par le biais de mutations sur l’ADNmt aura des conséquences sévères sur la survie des personnes porteuses de ces mutations. Par ailleurs, étant donnée la présence des mitochondries dans tous les types cellulaires de l’organisme, on peut également comprendre que plusieurs organes seront touchés simultanément. D’où la présentation hétérogène de ces maladies.
Les maladies mitochondriales sont transmises de plusieurs façons différentes.
- Soit, évidemment, par la mère et il s’agit là de la cause exclusive de thérapie par remplacement mitochondrial, puisque la maladie est prévisible.
- Soit du fait de mutations dites “sporadiques” qui n’étaient pas présentes chez la mère, et qui sont apparues pendant la vie fétale (ovogonies) ou dans l’ovocyte qui a donné naissance à l’enfant. Dans ces cas, la thérapie par remplacement mitochondrial est évidemment sans objet puisque la maladie n’était pas prévisible.
- Soit par des mutations dans des gènes nucléaires qui codent pour des protéines présentes dans la mitochondrie. Nous ne considèrerons pas ici ces maladies (également souvent sévères) car la thérapie par remplacement mitochondrial n’a évidemment aucun impact sur elles.
Les maladies mitochondriales les plus fréquentes sont les suivantes :
- Syndrome de Leigh: mort prématurée. Encéphalopathie, faiblesse musculaire, cardiomyopathie, troubles gastrointestinaux, détresse respiratoire. Mutation dans le gène ATPase (8993 T>G). Autres mutations d’expression tardives (complexes I, IV et V et ARN de transfert de lysine, valine et tryptophane). Transmission maternelle.
- MELAS : Myopathie, encéphalopathie, acidose lactique, convulsions. Mutation dans le gène ARNt-Leu (3243A>G). Autres mutations dans d’autres ARNt. Transmission maternelle.
- NARP: Neuropathie, ataxie, rétinite pigmentaire. Mutation dans le gène ATPase (8993 T>G). Hétéroplasmie 60-90%. Transmission maternelle.
- LHON: Neuropathie optique héréditaire de Leber. Trois mutations possibles dans les gènes formant le Complexe I, 11778 G > A, 3460 G > A, ou 144484 T > C. Perte de vision rapide (qq mois). Parfois réversible. Transmission maternelle.
- MERRF: Myoclonie, epilepsie, ataxie, fibres musculaires anormales. Mutation dans le gène ARNt-Lys (8344A > G, ou m.8356 T>C). Transmission maternelle.
- Déficience infantile en Cyt C Oxydase (COX): hypotonie et faiblesse musculaire sévères, détresse respiratoire. Mutation dans le gène ARNt-Glu (14674 T>C). Affecte le gène codant pour la COX. Curieusement, cette maladie est réversible entre 5 et 20 mois. Transmission maternelle.
- Syndrome de Pearson: mort prématurée. Anémie sidéroblastique (globules rouges; défaut incorporation haeme dans hémoglobine), dysfonction du pancréas, foie et rein. Large délétion dans l’ADNmt (ovocyte ou embryon). Transmission sporadique.
- KSS (Kearns-Sayre Syndrome): Rétinite pigmentaire, ophthalmoplégie externe progressive, atteintes cardiaque et cérébrale; apparaît avant 20 ans. Large délétion dans l’ADNmt (Complexes I, ATPase, COX). Transmission sporadique.
(Shapira 2012; Saneto 2013)
La thérapie par remplacement mitochondrial
Cette thérapie consiste à remplacer dans un ovule non fécondé ou dans l’embryon (zygote, stade à une cellule) les mitochondries porteuses de mutations par les mitochondries d’une femme non porteuse de mutation.
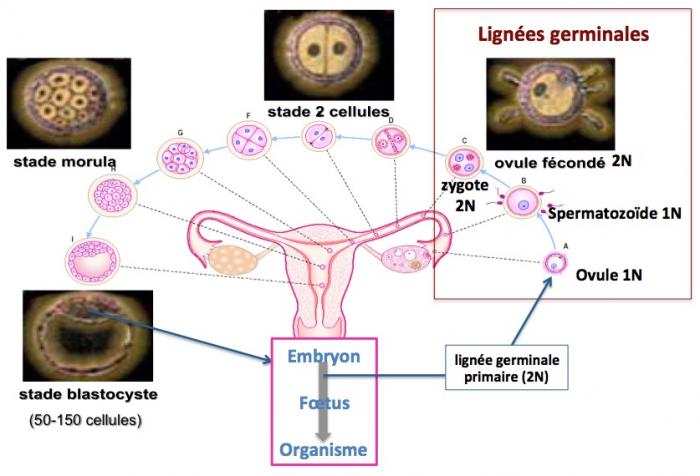
Déroulement normal de l'embryogénèse
Dans cette thérapie, la mère reste la mère biologique puisque son ADN nucléaire (chromosomes) est conservé.
Trois techniques de microchirurgie de transfert nucléaire peuvent être pratiquées :
- le transfert du karyoplaste (struture comprenant les chromosomes et le fuseau mitotique) dans l’ovocyte non fécondé.
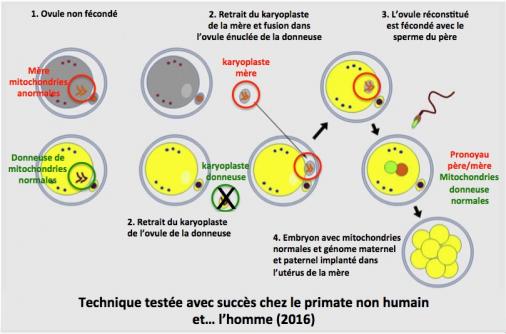
1. Des ovules non fécondés de la future mère (porteuse de mitochondries anormales) et de la donneuse de mitochondries normales sont recueillis.
2. Le karyoplaste de la donneuse est retiré et éliminé. Le karyoplaste de la mère est prélevé et introduit dans l’ovule énucléé de la donneuse.
3. L’ovule ainsi réconstitué est fécondé avec le sperme du père.
4. L’embryon qui contient maintenant les mitochondries normales et les chromosomes de la mère et du père est implanté dans l’utérus de la mère. (Amato 2014; Mitalipov 2014; Greefield 2014)
Il est possible qu’au cours du prélèvement du karyoplaste de l’ovule de la mère, une partie du cytoplasme soit également prélevée comme le montre la photo ci-dessous.
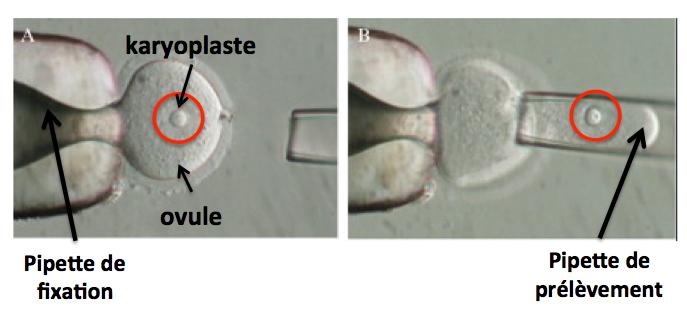
Ainsi, des mitochondries (en faible quantité) pourraient être introduites dans l’ovocyte énucléé de la donneuse et venir contaminer les mitochondries normales. Nous verrons les conséquences possibles de cette contamination plus loin.
- le transfert du pronoyau dans le zygote (embryon au stade une cellule).
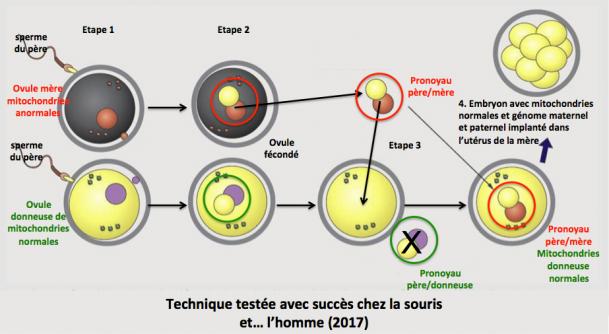
1. Des ovules de la future mère (porteuse de mitochondries anormales) et de la donneuse de mitochondries normales sont fécondés avec le sperme du père (zygote).
2. Le pronoyau des zygotes de la donneuse est retiré et éliminé.
3. Le pronoyau des zygotes de la mère est prélevé et introduit dans le zygote énucléé de la donneuse.
4. L’embryon qui contient maintenant les mitochondries normales et les chromosomes de la mère et du père est implanté dans l’utérus de la mère. (Amato 2014; Mitalipov 2014; Greefield 2014).
Il est possible qu’au cours du prélèvement du pronoyau des zygotes de la mère, une partie du cytoplasme soit également prélevée (comme indiqué plus haut au sujet du transfert du karyoplaste). Ainsi, des mitochondries (en faible quantité) pourraient être introduites dans le zygote énucléé de la donneuse et venir contaminer les mitochondries normales. Nous verrons les conséquences possibles de cette contamination plus loin.
- le transfert du globule polaire 1 dans l’ovocyte non fécondé.
Etant donné les risques de contamination mentionnés ci-dessus, une autre technique de transfert de mitochondrie est envisagée, le transfert du globule polaire 1. Mais avant de parler de cette technique, il nous faut préciser ce qu’est le globule polaire 1.
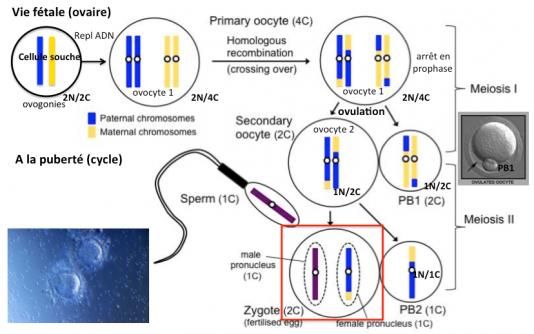
Les cellules de la lignée germinale (2N chromosomes, un provenant de la mère et un du père et 2C chromatides, une copie ADN de chaque chromosome) chez la femme sont générées au cours de l’embryogénèse (5eme semaine de sa vie fétale) et se différencient en ovogonies (2N,2C) qui répliquent leur ADN pendant la vie fétale pour donner les ovocytes primaires (2N,4C). Juste avant ou au moment de la naissance, les ovocytes primaires entrent en méiose et restent bloqués en prophase jusqu’à la pubertée (2N,4C). Au moment de l’ovulation, l’ovocyte primaire se divise de façon assymétrique pour donner un ovocyte secondaire (1N, 2C) qui contient l’essentiel du cytoplasme (mitochondries notamment) et le globule polaire 1 (1N,2C) qui, au contraire, contient très peu de cytoplasme. Lorsque l’ovocyte secondaire est fécondé par un spermatozoïde (1N, 1C) il entre en méiose (environ 2 heures après) pour donner le zygote (2N, 2C) et le globule polaire 2 (1N,1C).
Comme nous l’avons vu ci-dessus, l’ovulation de l’ovocyte primaire conduit à la formation du globule polaire 1. De nombreux travaux ont montré que ce globule polaire 1 possède un génome distinct de celui de l’ovocyte secondaire (du fait des recombinaisons génétiques) mais tout à fait normal. Il peut donc être utilisé pour réconstituer un ovule.
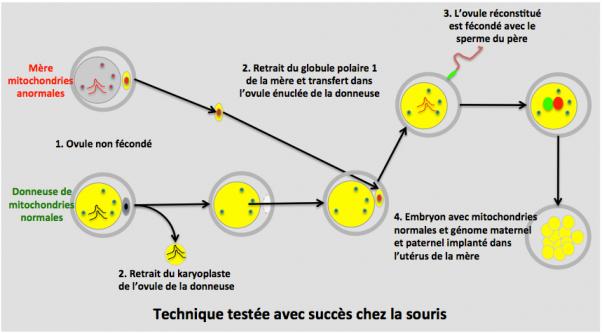
1. Des ovules non fécondés de la future mère (porteuse de mitochondries anormales) et de la donneuse de mitochondries normales sont recueillis.
2. Le karyoplaste de la donneuse est retiré et éliminé, ainsi que son GP1. Le GP1 de la mère est prélevé et introduit dans l’ovule énucléé de la donneuse.
3. L’ovule ainsi réconstitué est fécondé avec le sperme du père.
4. L’embryon qui contient maintenant les mitochondries normales et les chromosomes de la mère et du père est implanté dans l’utérus de la mère. (Greefield 2014; Ma 2017).
On voit ci-dessous que le prélèvement du GP1 est susceptible d’entraîner moins de cytoplasme et donc de mitochondries contaminantes que les prélèvement de karyoplaste ou de pronoyau.
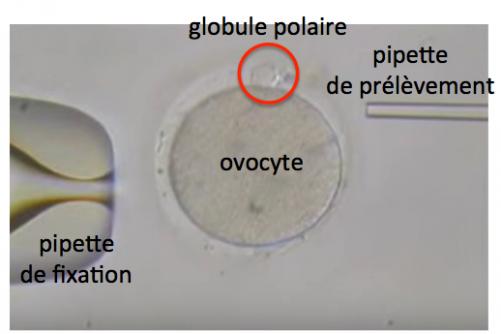
Premiers essais chez l’homme
Des essais concluants ont été réalisés chez des souris et des babouins.
Le premier essai chez l’homme a été réalisé au Mexique par l’équipe médicale Américaine du Dr John Zhang du New Hope Fertility Center, New York, durant l’été 2015. Du fait que l’autorisation de pratiquer cette intervention n’avait pas encore été accordée aux USA par la Food and Drug Administration (FAD), l’autorité qui statut sur les essais thérapeutiques, l’essai a eu lieu dans une clinique au Mexique (à Guadalajara) où il n’existe pas de réglementation sur ce type d’intervention. L’opération a été pratiquée selon les règles d’éthique approuvées au Royaume Uni, premier pays ayant ouvert sa réglementation à ce type de thérapie. Les parents étaient un couple Jordanien dont la mère était porteuse (saine) de la mutation sur le gène ATPase conduisant au syndrome de Leigh. Rappelons que cette maladie est mortelle et que deux enfants de ce couple étaient déjà décédés à l’âge de 6 ans et 8 mois. Les parents ont souhaité que le remplacement mitochondrial soit réalisé par transfert de karyoplaste car cette méthode évite la destruction d’un embryon. Cinq embryons ont été générés et un seul s’est développé normalement et a été implanté dans l’utérus de la mère (le niveau d’ADNmt muté était inférieur à 1%).
L’enfant (un garçon) est né le 6 avril 2016.
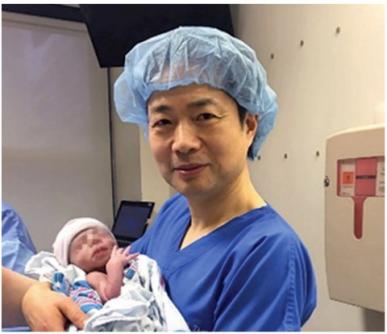
Dr John Zhang du New Hope Fertility Center
Un deuxième essai a été réalisé en Ukraine à Kyev par l’équipe du Dr Valery Zukin, de la Nadyia Clinic, au printemps 2016. Il n’existe pas de réglementation sur ce type d’intervention dans ce pays. Cette deuxième naissance soulève des questions d’éthiques. En effet, il s’agissait ici de corriger une infertilité par arrêt du développement embryoniaire du fait de mitochondries trop peu nombreuses, et non une maladie génétique. Le remplacement mitochondrial a été réalisé par transfert du pronoyau. L’enfant (une fille) est née le 5 janvier 2017. Elle est en bonne santé. Un autre enfant (un garçon) est né en Mars 2017. Il est en bonne santé (communication personnelle avec Valery Zukin, de la Nadyia Clinic).

La mère et l'enfant née le 5 Janvier 2017
Les risques biologiques
Les génomes nucléaire et mitochondrial ont évolué de concert depuis des centaines de millions d’années. Cette co-évolution a établi des interactions fonctionnelles entre les protéines issues de gènes nucléaires et les protéines issues des gènes mitochondriaux. En sera-t-il de même après transfert quand les gènes nucléaires et mitochondriaux proviendront de deux individus différents ?
 Lors du prélèvement du karyoplaste dans l’ovocyte de la mère ou du pronoyau dans le zygote issu de la fécondation de l’ovocyte de la mère, une (faible) partie du cytoplasme contenant les mitochondries mutées peut être entraînée et “contaminer” le nouvel embryon en s’y développant mieux que les mitochondries de la donneuse. Deux équipes ont récemment montré cette possibilité (Reinhardt 2013; Morrow 2015; Yamada 2016).
Lors du prélèvement du karyoplaste dans l’ovocyte de la mère ou du pronoyau dans le zygote issu de la fécondation de l’ovocyte de la mère, une (faible) partie du cytoplasme contenant les mitochondries mutées peut être entraînée et “contaminer” le nouvel embryon en s’y développant mieux que les mitochondries de la donneuse. Deux équipes ont récemment montré cette possibilité (Reinhardt 2013; Morrow 2015; Yamada 2016).
C’est la raison pour laquelle la “Human Fertilization and Embryology Authority” (HFEA) au Royaume uni et la FDA aux Etats Unis ont demandé une suspension des autorisations à pratiquer le remplacement mitochondrial jusqu’à ce que des études complémentaires soient réalisées sur ce problème.
Débats éthiques sur le remplacement mitochondrial
L’interdiction de modifier le génome des lignées germinales a été acté par la Convention sur les Droits de l’Homme et la Biomédecine (non signée par le Royaume Uni) et acceptée en 1997 par une partie de la communauté internationale et la Communauté Européenne.
L’article 13 de cette convention stipule que : “an intervention seeking to modify the human genome may only be undertaken for preventive, diagnostic or therapeutic purposes and only if its aim is not to introduce any modification in the genome of any descendants” (toute intervention ayant pour but de modifier le génome humain devra être entreprise seulement à des fins préventives, diagnostiques ou thérapeutiques, et seulement si son but final est de n’introduire aucune modification dans le génome d’aucun membre de la descendance).
En accordant son autorisation à la thérapie par remplacement mitochondrial, le gouvernement britannique s’est appuié sur le fait que cette thérapie ne modifie pas le génome nucléaire qui est le seul à définir les caractéristiques propres à l’individu (forme, traits et caractères individuels). De plus, l’autorisation a restreint la thérapie aux embryons filles. Dans un premier temps seuls des enfants males seront ainsi générés pour éviter tout risque de propagation à leur descendance si des problèmes encore inconnus devaient survenir sur le long-terme. La FDA (USA) a d’abord contesté les vues britanniques sur le fait que la thérapie par remplacement mitochondrial est une forme de modification du génome et a rejeté l’idée qu’il s’agissait d’un traitement. Dans un deuxième temps, la FDA accepta la possibilité de délivrer une autorisation après des études et une démarche conduites avec une “extrême prudence”. En France, et pour l'heure, seule la recherche sur le remplacement mitochondrial est autorisée.
Voici enfin une liste (non exhaustive) des questions éthiques que pose la thérapie par remplacement mitochondrial (Appelby 2017; Liao 2017, Baylis 2017, Scully 2017; Newson & Wrigley 2017) :
- Cette thérapie affectera-t-elle l’identité de l’enfant et si oui, comment ?
- Crée-t-elle un nouvel individu ou altère-t-elle les caractéristiques de l’individu ?
- Cette thérapie affecte-t-elle l’identité numérique* ou l’identité qualitative** ? Pour la HFEA : seule l’identité qualitative est affectée (uniquement la production d’énergie des cellules). Cependant selon le Nuffield Council of Bioethics : l’identité numérique est affectée; il existe en effet une différence par rapport à l’individu qui n’aurait pas subit cette intervention puisqu’il n’existerait pas!
- Cette thérapie présente un risque de dérive eugénique. Commentaire de P Maurel: le diagnostic préimplantatoire dans le cas de suspicion de maladie génétique sévère est accessible aux couples concernés depuis 1994. Cette "dérive" eugénique existe donc déjà.
- Filiation génétique: est-ce réellement une nécessité ou “seulement” un désir de la mère ?
- Il existe d’autres moyens d’avoir un enfant : adoption, don d’ovule, don d’embryon, et le diagnostic préimplantatoire n’est pas totalement fiable.
- Les enfants issus de cette thérapie seront “non naturels”. Quel en sera l’impact sur leur sens du Moi ?
- Cette thérapie est un moyen de sélection plus qu’une thérapie. Commentaire de P Maurel: le diagnostic préimplantatoire déjà autorisé (voir ci-dessus) est aussi un moyen de sélection.
- Il s’agit d’une technologie coûteuse au regard des autres besoins de l’humanité.
* identité numérique : moi (PM) et la personne qui écrit ce texte en ce moment sur mon mac
** identité qualitative : deux objets fabriqués en série sont qualitativement identiques
En résumé
- Les mitochondries jouent un rôle essentiel dans la vie des cellules des eukaryotes
- Elles sont uniquement transmises par la mère
- Elles possèdent leur propre génome
- Leur génome est sujet à des mutations comme le génome nucléaire
- Ces mutations peuvent conduire à des maladies génétiques graves, fortement invalidantes et parfois mortelles
- Le diagnostic de transmission de ces maladies est très difficile (du fait de l’hétéroplasmie)
- Un moyen de les guérir consiste à remplacer les mitochondries anormales de la mère par celles d’une donneuse de mitochondries normales
- La thérapie par remplacement mitochondrial est techniquement possible et préserve la "maternité génétique" de la mère
- Cette technique présente cependant des risques médicaux (adéquation entre génome nucléaire et mitochondrial) et pose des questions éthiques
- Déjà deux enfants sont nés (peut-être trois)
- à suivre…
Bibliographie
Ce dossier a été rédigé sur la base d'articles scientifiques publiés dans des revues internationales à comité de lecture.
Dans la liste ci-dessous, un clic sur "PubMed" permet d'ouvrir le résumé de l'article.
L'astérisque (*) devant le nom de l'auteur indique que la version complète de l'article est accessible à tout public sur le site du journal
*Amato et al. 2014. PubMed
Greenfield, 2014. Medical Research Council Harwell and HFEA
Gustafsson et al. 2016. PubMed
*Koonin 2015. PubMed
Lightowlers et al. 2015. PubMed
Ma et al. 2017. PubMed
Mandavilli et al. 2002 PubMed
*Margulis et al. 2006. PubMed
*Mitalipov et al. 2014. PubMed
*Morrow et al. 2015. PubMed
*O'Malley et al. 2015. PubMed
Picard et al. 2016. PubMed
*Protti et al. 2006. PubMed
Reinhardt et al. 2013. PubMed
*Saneto et al. 2013. PubMed
Schapira 2012. PubMed
Stewart et al. 2015. PubMed
Taylor et al. 2005. PubMed
Vafai et al. 2012. PubMed
*Wallace 2015. PubMed
*Wang et al. 2014. PubMed
Yamada et al. 2016. PubMed
Zheng et al. 2006. PubMed
Appelby et al. 2017. Bioethics. Special issue. The ethics of mitochondrial replacement 2017
Baylis et al. 2017. Bioethics. Special issue. The ethics of mitochondrial replacement 2017
Liao et al. 2017. Bioethics. Special issue. The ethics of mitochondrial replacement 2017
Newson et al. 2017. Bioethics. Special issue. The ethics of mitochondrial replacement 2017
Scully et al. 2017. Bioethics. Special issue. The ethics of mitochondrial replacement 2017
Date de dernière mise à jour : 13/05/2023
Ajouter un commentaire