
Les variants du SARS-CoV-2
Résumé tout public
Les discussions sur les variants du SARS-CoV-2, le virus responsable de la COVID-19, sont omniprésentes dans les médias et à juste titre. Le but de cette page est d'expliquer ce que sont ces variants, comment ils apparaissent, quels risques ils représentent, et comment les combattre.
Préambule
On ne peut évoquer les variants du SARS-CoV-2 sans parler du terrible combat qui se livre en permanence et sans la moindre concession au sein de notre organisme.
Les belligérants : le virus et le système immunitaire.
Le champ de bataille : nos cellules.
Le virus tente d'infecter et d'utiliser les ressources de nos cellules afin de produire ses descendants, appelés les virions.
Le système immunitaire traque sans relâche et détruit le plus grand nombre de virions ou de cellules infectées. Dans ce combat pour la vie chaque adversaire tente de surpasser l'autre.
Leurs armes : le camouflage pour le virus, les anticorps et les lymphocytes T cytotoxiques pour le système immunitaire.
Chez les virus, le camouflage consiste d'une part à bloquer ou affaiblir l'activité du système immunitaire afin de lui échapper et d'autre part à se reproduire sous forme de nouveaux virions par le biais de mutations dans leur génome. Les mutations sont des erreurs commises par les protéines virales lorsqu'elles recopient le génome pour transférer celui-ci aux nouveaux virions. Grâce aux modifications qu'entraînent les mutations sur ces virions, ils est possible qu'ils échappent aux anticorps produits par le système immunitaire.
Le SARS-CoV-2 ne faut (faillir) pas à cette règle.
Approximativement et en moyenne, une erreur apparaît dans le génome du SARS-CoV-2 chaque fois que 30 nouveaux virions sont produits, et on estime qu'entre 10 à 100 millions de nouveaux virions sont produits chez un malade lors du pic infectieux. En fait, les mutations qui affectent le génome des virions constituent un moyen extrêmement efficace d'adaptation aux conditions hostiles qu'ils rencontrent dans les cellules de l'hôte, et permettent au virus notamment d'échappemer au système immunitaire.
C'est quoi au juste un variant ?
A partir d'un virion donné (considéré comme "ancêtre"), les mutations qui apparaissent dans le génome des virions émergeants peuvent procurer à ces derniers des propriétés différentes de celles de leur ancêtre en termes par exemple de contagiosité, de vitesse de réplication, de pathogénicité, de résistance aux anticorps, etc. Ces nouvelles propriétés peuvent conférer au nouveau virion un avantage ou un désavantage pour sa survie par rapport à son ancêtre et/ou à ses congénères.
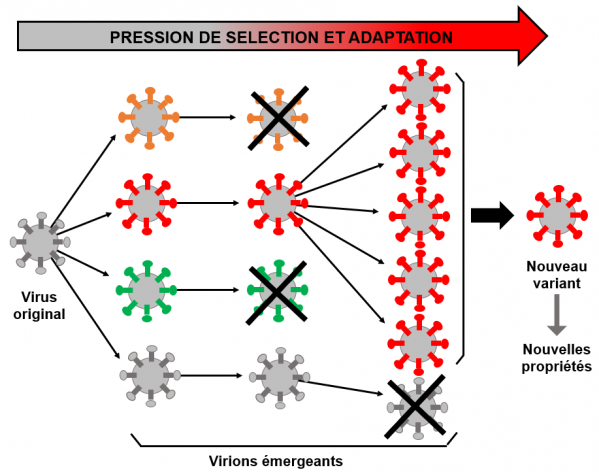
Naissance d'un variant (schéma très simplifié)
Dans ce schéma, seule la protéine S du virus est considérée. A gauche le virus "original (protéine S de couleur grise). Dans les cellules du malade infecté, ce virus (gris) se réplique et produit des virions (virions émergeants) dans lesquels des mutations peuvent apparaître. Ces mutations sont représentées ici par des virions de couleurs différentes. Certains des virions émergeants (orange et vert) ne survivent pas pour diverses raisons (anticorps, compétition avec les autres virions,etc.). Certains au contraire prospèrent (virion rouge) jusqu'à devenir une population majoritaire bien adaptée. D'autres virions (gris) disparaissent par exemple du fait de la compétition avec les virions rouges. Il apparaît alors un nouveau variant. Ce type d'évolution des populations virales est suspecté se produire chez des personnes infectées dont le système immunitaire est déficient et chez qui l'infection devient chronique (voir dans la suite du texte). Le variant peut alors être propagé par le malade et, de proche en proche, à d'autres personnes.
Si un virion acquiert grâce à une ou plusieurs mutations un avantage important sur ses congénères (par exemple une reproduction plus rapide, une meilleure résistance aux anticorps, etc.) il va prendre le dessus au gré des cycles de reproduction et engendrer une population importante de virions possédant le même génome. Si cet ensemble de virions prolifère et survit, il entre en compétition avec les autres virions émergeants et peut atteindre un nombre d'individus suffisant pour devenir une population majoritaire du virus. Il est alors considéré comme un variant. Ce variant peut alors être transmis par le malade à d'autres personnes qui le transmettront à leur tour.
Le variant représente donc un large ensemble de virions qui possèdent tous un génome identique ou très proche mais suffisamment différent de celui du virus "original" pour se comporter différemment à divers niveaux, contagiosité, vitesse de prolifération, mécanisme de pathogénèse, résistance au système immunitaire, etc.
Chez le SARS-CoV-2, le gène qui code pour la protéine S est dit "hypervariable". C'est à dire qu'il accumule les mutations.
Pourquoi ?
Parce que cette protéine est la cible principale du système immunitaire.
La protéine S joue un rôle majeur dans le processus infectieux. C'est par elle que le virus s'accroche à son récepteur sur les cellules, première étape de l'infection (voir sur ce site la page "Entrée du virus dans les cellules"). De fait, elle devient la cible principale dans le "viseur" du système immunitaire. D'où l'intérêt pour le virus de la modifier par accumulation de mutations afin qu'elle ne soit plus aussi bien reconnue (camouflage).
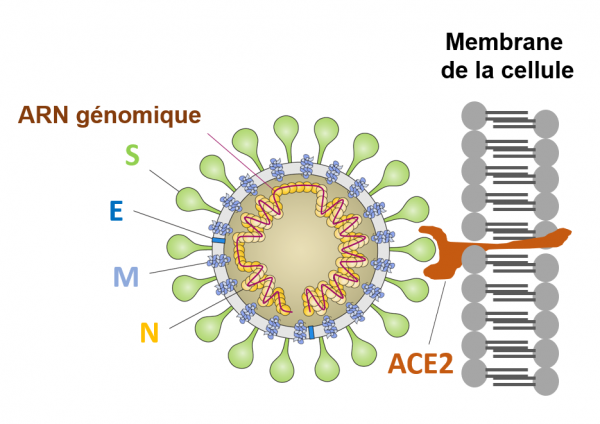
Protéine S et ACE2
Le virus est schématisé ici sous forme d'une sphère constituée d'une membrane lipidique en gris clair, sur laquelle sont implantées diverses protéines, la protéine S (spike), la protéine E (enveloppe), la protéine M (membrane). A l'intérieur de la membrane virale on trouvent l'ARN génomique (ruban de couleur marron, qui code pour l'ensemble des protéines du virus) et la protéine N (nucléocapside). Cette protéine N sert à empaqueter l'ARN viral. A côté des protéines S, E, M et N, le génome viral code pour une vingtaine d'autres protéines qui sont produites par la machinerie cellulaire après que le virus soit entré dans les cellules et y ait libéré son ARN. Le schéma montre la liaison entre la protéine S du virus et son récepteur cellulaire, la protéine ACE2 qui se trouve sur la partie externe des membranes cellulaires. Cette liaison est la première (et déterminante) étape du processus d'entrée du virus dans les cellules.
Le système immunitaire quant à lui patrouille et traque sans relâche les différents virions en adaptant sa fabrication d'anticorps à chaque nouvel arrivant. Mais cela prend du temps.
Est-ce un combat sans fin ?
Oui et non. Tout dépend de la rapidité d'adaptation et de réaction des deux adversaires. Si le virus mute très rapidement, il réussira à faire émerger un nombre énorme de nouveaux virions avant que le système immunitaire ait eu le temps de produire des anticorps qui leurs seront adaptés. Dans ce cas le système immunitaire finira par être dépassé et la maladie virale pourra s'installer. C'est le cas des virus comme le virus du SIDA (VIH) ou de l'hépatite C (VHC) contre lesquels jusqu'ici aucun vaccin efficace n'a pu être produit. Ces maladies sont par contre guéries par polythérapie pharmacologique.
Si au contraire, le virus mute lentement, alors le système immunitaire aura le temps de produire des anticorps spécifiques et finira pas éradiquer les nouveaux varions et donc l'infection virale.
Par chance, le coronavirus SARS-CoV-2 responsable de la pandémie actuelle est un virus qui mute relativement lentement. Cela ne signifie pas pour autant que ce virus soit éradiqué par le système immunitaire sans problème et la pandémie que nous vivons nous le montre tous les jours. De plus, la présence des variants en est une preuve flagrante. Ces variants sont le signe que le virus est capable de s'adapter en permanence à la contrainte qu'il reçoit du système immunitaire en cherchant à se camoufler au mieux et notamment en mutant.
A ce stade, il faut prendre en compte l'arrivée de renforts pour le système immunitaire. Et ces renforts sont les vaccins. Les vaccins agissent comme des instructeurs qui apprennent au système immunitaire comment combattre au mieux les virions en lui désignant les cibles les plus sensibles ou les plus stratégiques qu'il va devoir absolument viser.
Les variants d'intérêt
A l'heure actuelle, mais c'est une situation qui va changer à plus ou moins brève échéance, il existe au moins cinq variants principaux dits "d'intérêt", du fait des préoccupations qu'ils provoquent.
Le premier est le variant "Britannique" découvert dans le sud de l'Angleterre en Septembre 2020. Ce variant comporte plusieurs mutations par rapport au virus "original" (celui découvert à Wuhan en Novembre 2019). Une d'entre elles lui confère un avantage important. Cette mutation, apparue dans la séquence de la protéine S, renforce l'interaction entre cette protéine et le récepteur ACE2 situé à la surface des cellules. De fait, le variant pénètre plus rapidement dans les cellules et manifeste donc une contagiosité accrue.
Les deux autres variants Sud-Africain et Brésilien, ont été découverts respectivement dans la région de Mandela Bay à l'est du Cap en Août 2020, et dans la région de Manaus en Amazonie en Décembre 2020. Ces deux variants comportent plusieurs mutations et notamment la mutation trouvée sur la protéine S dans le variant Britannique, qui leur confère donc une contagiosité accrue. Cependant en plus de cette mutation, ils en comportent deux autres sur la protéine S qui contribuent à diminuer la liaison entre les anticorps anti-SARS-CoV-2 et la protéine S, conférant ainsi à ces variants la propriété de résister au système immunitaire naturel et aux vaccins qui ciblent justement la protéine S du virus.
Les variants Californien et New-Yorkais. Le variant Californien a été observé pour la première fois en juillet 2020 par des chercheurs du Cedars-Sinai Medical Center, en Californie à Los Angeles. Il est resté discret durant l'été 2020, mais est réapparu toujours en Californie en Novembre 2020, date à partir de laquelle son incidence n'a cessé d'augmenter, passant de 36 % en Novembre à 50% en Janvier 2021. Le variant New-Yorkais avait été observé une première fois en Novembre 2020 dans les départements de microbiologie et de maladies infectieuses de l'Université Columbia à New-York. Par la suite, il a été constaté une augmentation substantielle des cas positifs pour ce variant au fil du temps, de 1,3 % en Novembre 2020 à 5,3 % à la mi-Janvier, et finalement à 12,3 % entre le 8 et le 15 Février 2021. Ces variants comportent plusieurs mutations sur la protéine S (différentes de celles des variants ci-dessus) qui se sont avérées conférer non seulement une contagiosité accrue et un échappement aux anticorps, mais également pourrait produire une COVID-19 plus sévère mais cela reste à démontrer.
Le dernier venu dans cette série est le variant Indien. Alors que le nombre de cas de coronavirus en Inde a augmenté à partir d'avril 2021, des millions de personnes ont convergé vers le Gange pour se baigner dans un lieu sacré offrant une chance de salut. Lorsque les pèlerins sont rentrés chez eux dans tout le pays, certains ont apporté le virus avec eux. Au pic de l'infection (mi-mai 2021), ce variant a produit plus de 400 000 cas de COVID-19 par jour avec plus de 4500 morts par jour.
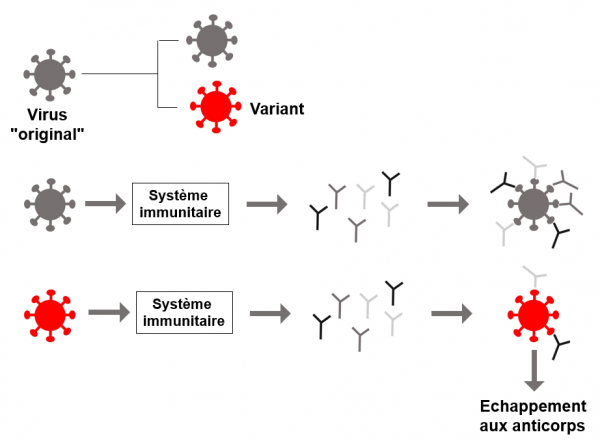
Résistance d'un variant au système immunitaire (schéma très simplifié)
On a vu plus haut comment un variant s'installe à partir du virus "original". Au moment de l'émergence du variant le système immunitaire a généré des anticorps et des lymphocytes T cytotoxiques qui sont adaptés à combattre le virus "original". Les nouvelles propriétés acquises par le variant (notamment les mutations affectant la protéine S, cible principale des anticorps), font que le nombre des anticorps capables de cibler la nouvelle protéine S du variant est fortement diminué (seulement 2 sur 6 dans le schéma présenté). De fait, le variant peut survivre.
Hormis la mise en place de mesures limitant les interactions physiques avec les populations affectées au premier chef par ces variants (restrictions des entrées sur le territoire, mise en quarantaine des personnes testées positives et des cas contacts, etc.), il n'existe pour le moment pas de barrière totalement efficace contre ces variants. Les problèmes que posent les variants Sud-Africain et Brésilien sont particulièrement préoccupants puisqu'ils font passer l'efficacité des vaccins Pfizer et Moderna de 95% (avec le virus "original") à 50%, et celle du vaccin AstraZeneka de 70-90% à 20% avec le variant Sud-Africain. Il en est de même pour le variant Indien qui réduit l'efficacité des vaccins à 88% pour le Pfizer et 60% pour l'AstraZeneca.
Le problème de cette limitation d'efficacité des vaccins (qui ont été conçus pour le virus "original") a déjà été appréhendé par les fabricants de vaccins. Ceux-ci travaillent déjà sur les nouvelles versions de leur vaccin qui seront dirigées spécifiquement contre ces variants. Dans cette optique, les vaccins à ARNm et les vaccins adénoviraux à réplication défectueuse peuvent être adaptés assez rapidement pour tenir compte des changements de séquence spécifiques (et déterminants) présents dans les nouveaux variants.
En conclusion, la formation de variants dans les maladies virales est un phénomène naturel et inéluctable. A côté des variants d'intérêt décrit ci-dessus, l'émergence d'autres variants se profile à l'horizon (avec notamment un nouveau variant Américain) dont je reparlerai plus tard.
Pour celles et ceux qui souhaitent approfondir leurs connaissances sur ce sujet, voir la suite ci-dessous.
Les variants du SARS-CoV-2
La survie à long terme de toute les espèces vivantes (bactéries, végétaux, animaux, homme) est basée sur leur capacité à se reproduire en maintenant et transmettant leur génome (ADN), en théorie à l'identique. Comme tout être vivant, la destinée d'un virus est de se reproduire. Les virus sont de parfaits parasites, incapables de produire leurs propres protéines et donc de se reproduire en dehors d'une cellule bactérienne, végétale, animale ou humaine. La reproduction virale - qui n'est pas sexuée - se fait à l'intérieur des cellules infectées. Dans le cas du SARS-CoV-2 il s'agit principalement des pneumocytes de type 2 (cellules qui tapissent les alvéoles pulmonaires). Ces cellules fournissent au virus le matériel nécessaire à sa réplication et notamment assurent la synthèse de ses protéines. A partir de là, le virus peut se reproduire en utilisant ses propres protéines pour produire et recopier son génome afin d'assembler de nouveaux virions qui vont quitter la cellule pour en infecter d'autres.
Et c'est là que des différences importantes existent entre les végétaux, animaux ou humains et les virus.
Chez l'homme par exemple, la synthèse des multiples copies du génome (ADN constitué de 3,4 milliards de nucléotides, adénine, thymine, guanine et cytidine, figurés par les quatre lettres A, T, G, et C) est très "fidèle" avec environ une erreur chaque 10 milliards de nucléotides synthétisés. C'est à dire que l'ensemble du génome (3,4 milliards de lettres) est reproduit sinon à l'identique, du moins avec seulement 0,3 erreur par copie. Sans entrer dans les détails, cette "fidélité" tient au fait qu'il existe chez l'homme (et les autres espèces végétales et animales) des systèmes de relecture et de correction de l'ADN extrêmement performants (Jiang et al., Ann Transl Med 2020). Chez les coronavirus, au contraire, la synthèse du génome (ARN) est "infidèle" avec environ une erreur chaque million de nucléotides synthétisés (Smith et al. Ann Rev Virol 2014; Sanjuan et al., Cell Mol Life Sci 2016). C'est à dire qu'approximativement et en moyenne une erreur apparaît dans le génome du SARS-CoV-2 chaque fois que 30 virions sont produits. Cette "infidélité" tient au fait que les systèmes de relecture et de correction sont inexistants ou peu performants.
Un exemple très simple est le suivant. Vous devez recopier un texte. Si vous êtes très attentif et si vous relisez le texte que vous avez écrit, il y aura peu (ou pas) d'erreurs. Si au contraire vous êtes étourdi ou peu attentif, et si vous négligez de relire le texte que vous avez écrit, il y aura beaucoup d'erreurs. C'est exactement ce qui se passe quand on compare les végétaux ou animaux et les virus.
On estime qu'à tout moment il existe environ 10 virions SARS-CoV-2 infectieux à l'intérieur d'une cellule, et qu'une cellule infectée produit entre 10 et 100 virions infectieux sur quelques jours. De plus, on estime qu'environ un million de cellules pulmonaires sont infectées au moment du pic infectieux chez un malade (Sender et al. MedRxiv 2020). On doit donc s'attendre approximativement à une production de 10 à 100 millions de nouveaux virions chez un malade. D'où l'apparition de variants. Un dernier point à noter est le suivant. Parmi toutes les mutations enregistrées depuis le début de la pandémie, la plupart sont dites silencieuses (ou "synonymes"), c'est à dire qu'elles ne conduisent pas à des modifications au niveau des protéines virales. Seules les mutations dites "non-synonymes" concourent à produire des variants qui manifestent de nouvelles propriétés (contagiosité augmentée, échappement aux anticorps, etc). Voir à ce sujet, le paragraphe ci-dessous sur le code génétique.
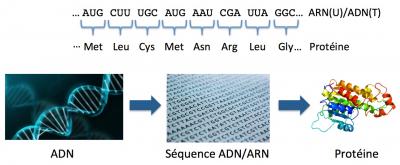
Le code génétique (rappel)
Le code génétique permet de convertir une séquence d’ADN ou d’ARN (4 nucléotides) en protéine (20 acides aminés). A chaque triplé de bases nucléotidiques (A, T/U, G ou C) correspond un acide aminé. On notera que dans ce code, le nucléotide T (présent dans l'ADN) est remplacé par le U dans l'ARN qui est traduit en protéine par les ribosomes. Il existe 64 triplets possibles à partir de 4 nucléotides. Cela signifie que plusieurs triplets de nucléotides codent pour un même acide aminé, comme indiqué dans la figure ci-dessous. On peut voir également que plusieurs triplets (UAA, UAG et UGA) correspondent à un STOP, c’est à dire qu’ils indiquent au ribosome l’arrêt de la traduction en protéine.
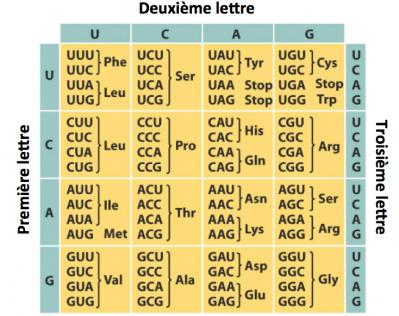
Le code génétique
Mutations synonymes et non-synonymes. Exemple d'une mutation synonyme. On voit dans le tableau que par exemple pour l'acide aminé Leucine (Leu, première colonne) il existe 4 codons possibles: CUU, CUC, CUA, CUG. Cela signifie que toute modification qui pourrait intervenir sur le nucléotide en position 3 du codon ne changera pas la nature de l'acide aminé (Leu) et la protéine sera inchangée. La mutation est dite synonyme. Exemple d'une mutation non-synonyme. Prenons l'acide aminé Isoleucine (Ile) première colonne du tableau, codée par les codons AUU, AUC et AUA. Si une mutation touche le nucléotide en position 2 par exemple AUU changé en ACU, l'acide aminé isoleucine est changé en thréonine (Thr) et on doit donc s'attendre à une modification de la protéine. La mutation est dite non-synonyme, puisqu'elle affecte la séquence de la protéine qui sera traduite. A côté de ce type de mutation, il existe d'autres possibilités de mutations comme par exemple celles qui conduisent à un STOP (UAA, UAG, UGA), ou à la perte de nucléotides (délétions).
Pourquoi les variants acquièrent-ils de nouvelles propriétés ?
Nous avons vu ci-dessus que les variants comportent des mutations dans leur génome par rapport au génome du virus "original", celui découvert à Wuhan fin 2019 (Wu et al. Nature 2020). Or, ces mutations dans l'ARN viral peuvent dans certains cas (mais pas toujours, voir ci-dessus le paragraphe sur le code génétique) avoir pour conséquence des modifications (mutations) de la séquence en acides aminés des protéines du virus. C'est ce type de mutation qui entraîne l'apparition de propriétés nouvelles pour le variant en question. Les variants sont donc des virus qui possèdent les mêmes caractéristiques générales que le virus "original", mais au gré des mutations ils peuvent acquérir des propriétés différentes : meilleure capacité d'infection, prolifération plus rapide, résistance aux anticorps, etc. A noter que les mutations représentent un moyen pour le virus de se camoufler et de survivre dans un environnement hostile (anticorps, notamment).
Pourquoi se focalise-t-on sur la protéine S (Spike) des variants du SARS-CoV-2 ?
Le génome du virus SARS-CoV-2 code pour plus de 25 protéines différentes. Alors pourquoi se focaliser sur la protéine S ?
Deux raisons.
Premièrement, c'est la protéine S qui "ouvre la porte" de nos cellules au virus. Cette protéine S se fixe sur une protéine présente sur la membrane extérieure des cellules pulmonaires, appelée ACE2. On dit que ACE2 est le récepteur du virus. L'accrochage de S sur ACE2 entraîne le processus d'entrée du virus dans les cellules (voir sur ce site ICI). C'est donc une étape essentielle dans le processus d'infection. Par exemple, la souris sauvage est totalement insensible au SARS-CoV-2 parce que la protéine S du virus ne peut s'accrocher sur sa protéine ACE2, à l'inverse de la chauve-souris, du pangolin ou du hamster Syrien par exemple.
Deuxièmement, l'analyse des anticorps présents dans le sang des personnes qui ont contracté la COVID-19 montre que l'immense majorité des anticorps ciblent la protéine S du virus et très peu d'entre eux ciblent d'autres protéines (Piccoli et al., Cell 2020; Barnes et al. Cell 2020). Cela signifie que la protéine S figure parmi les protéines virales les plus immunogènes. Cela confirme aussi qu'elle joue un rôle important dans l'infection. Cependant, on ne peut exclure le fait que des mutations dans d'autres protéines virales qui se produisent également influent sur les propriétés des nouveaux variants.
Or, on a retrouvé entre 8 et 10 mutations dans la protéine S des variants qui circulent en ce moment. En particulier certaines de ces mutations se retrouvent précisément dans la partie de la protéine S qui assure la liaison sur le récepteur ACE2. Il s'avère que cette partie du génome viral est une région hypervariable, c'est à dire, sujette à une intense production de mutations.
Quels sont les principaux variants d'intérêt connus à l'heure actuelle, et quels risques représentent-ils ?
A côté du virus SARS-CoV-2 "original", c'est à dire celui apparu à Wuhan fin 2019 (Wu et al. Nature 2020), plusieurs variants sont maintenant recensés. Certains de ces variants sont dits "d'intérêt" car ils posent de sérieux problèmes en tant qu'éléments aggravants de la pandémie.
Le premier variant (G614)
Ce variant est le premier apparu en Chine au début de la pandémie entre Février et Mars 2020. Il comporte une mutation (D614G) dans la protéine S, associée à 3 autres mutations réparties sur l'ensemble du génome (Hou et al., Science 2020; Plante et al. 2020; Tang et al., J Infect 2021).
A noter: la mutation "D614G" signifie que l'acide aminé D (acide aspartique, dans le code à une lettre) en position 614 de la protéine S (qui compte au total 1273 acides aminés) est changé par l'acide aminé G (glycine). Ce mode de notation des mutations est utilisé tout au long de cette page.

Séquence de référence de la protéine S (spike) dans le virus "original" de Wuhan
La séquence est représentée ici (banque de données du National Center for Biotechnology Information; et Wu et al. Nature 2020) selon le code des acides aminés à une lettre: Alanine A, Arginine R, Acide Aspartique D, Asparagine N, Cysteine C, Acide Glutamique E, Glutamine Q, Glycine G, Histide H, Isoleucine I, Leucine L, Lysine K, Methionine M, Phenylalanine F, Proline P, Serine S, Threonine T, Tryptophane W, Tyrosine Y, et Valine V. La partie soulignée représente le domaine de liaison de la protéine S au récepteur ACE2. La partie soulignée en gras représente le motif (le coeur) de liaison de la protéine S au récepteur ACE2. La protéine S comporte 1273 acides aminés.
La fréquence de cet ensemble de mutations a augmenté non seulement au sein de régions géographiquement distinctes pendant l'épidémie, mais également au niveau mondial, ce qui suggère que l'émergence de ce variant était le résultat d'un avantage adaptatif du virus plutôt que d'un effet fondateur (transmission du virus à plusieurs personnes à partir d'un unique individu porteur original du variant) et/ou d'une dérive génétique (mutations au hasard). Ce variant, appelé G614, s'est rapidement transmis à l'ensemble de la planète. Alors qu'il était encore minoritaire avant Mars 2020, il est devenu le variant principal au niveau mondial avec une prévalence de 74% en Juin 2020. Ce variant manifeste une augmentation de sa réplication et une stabilité supérieure dans les tissus primaires des voies respiratoires. Cette mutation confère aux variantes la capacité de se propager plus rapidement que le virus "original" (Hou, Science 2020; Plante, Nature 2020). Il existe également des preuves épidémiologiques que les variants présentant cette mutation spécifique se propagent plus rapidement que les virus ne présentant pas cette mutation (Volz et al., Cell 2021; Korber et al., Cell 2020). Par contre le variant G614 ne semble pas augmenter la mortalité des personnes atteintes. Bien que ce variant n'échappe pas aux anticorps neutralisants, il constituait déjà un avertissement sans frais sur le fait que cette situation pouvait se produire. Enfin, les 3 variants décrits ci-dessous descendent de celui-ci car ils comportent tous la mutation D614G.
Le variant B.1.1.7 selon la nomenclature "Pangolin" (dit variant "Britannique").
A noter: la nomenclature Pangolin est utilisée tout au long de cette page pour désigner les variants. Cette nomenclature n'a strictement rien à voir avec le coronavirus du pangolin (l'animal). Il s'agit d'un acronyme signifiant : Phylogenetic Assignment of Named Global Outbreak LINeages (Rambaut et al., Nature 2020). Ce mode de nomenclature est basé sur l'arbre phylogénétique (généalogique) des variants déduit du séquençage de leur génome et de la comparaison de ces divers génomes.

Séquence de la protéine S (spike) du variant Britannique
La séquence est représentée ici (banque de données du National Center for Biotechnology Information; et Rambaut et al. Virological 2021) selon le code des acides aminés à une lettre. Les mutations par rapport à la séquence de la protéine S du virus "original" sont en rouge surilignées en jaune.
Ce variant a été découvert dans le Kent le 20 Septembre 2020 (https://cov-lineages.org/global_report_B.1.1.7.html). Il s'est tout d'abord manifesté par le fait que dans les résultats des tests RT-PCR commercialisés par Thermo-Fisher (largement utilisé au Royaume Uni) qui ciblent 3 gènes viraux orf1ab, S et N, le gène codant pour la protéine S était négatif alors que les 2 autres étaient positifs. Il s'est avéré que ce résultats était dû à une mutation de ce gène dans le variant (conduisant à la la délétion 69-70 dans la protéines S) (Davies et al., MedRxiv 2021). La négativité des tests PCR pour le gène S a donc été utilisée dans un premier temps pour détecter ce variant (S-gene target failure, SGTF). Sa présence a ensuite été confirmée par séquençage systématique (Rambaut, Virological 2021). Ce variant comporte 17 mutations sur l'ensemble du génome dont 8 touchent la protéine S avec une mutation (N501Y) dans le domaine de liaison de cette protéine au récepteur ACE2. En moins de deux mois, ce variant a véritablement envahi le Royaume-Uni entre le 12 octobre où sa prévalence était de 3% et fin décembre où il est devenu le variant majoritaire (Public Health England, Technical briefing 14 January 2021). De plus, ce variant est maintenant présent dans plus de 50 pays dont la France (avec 39 cas au 12 Janvier 2021, selon le Conseil Scientifique COVID-19) et le Portugal où il est en train de provoquer un regain de l'épidémie. Ce variant manifeste une contagiosité supérieure à celle du SARS-CoV-2 "original" (R0 voisin de 1,5). Ce regain de contagiosité semble lié à la mutation N501Y (Tang et al., J Infect 2021). Quant à sa léthalité il semblerait selon une étude Anglaise toute récente qu'elle pourrait être légèrement supérieure à celle liée au SARS-CoV-2 "original". La probabilité de mourir de la COVID due à ce variant augmenterait d'environ 64% selon les derniers résultats (Challen et al. BMJ2021). En ce qui concerne l'impact de ce variant sur l'efficacité des vaccins, il semble heureusement qu'il n'y ait pas d'interférence. Cependant, la présence d'une nouvelle mutation E484K vient d'être détectée chez 11 personnes non apparentées porteuses de ce variant (Public Health England, Technical briefing 14 January 2021). Or cette mutation, présente dans les variants Sud-Africain et Brésilien, semble impliquée dans la résistance du virus aux anticorps naturels et aux vaccins (voir les paragraphes ci-dessous).
Le variant B.1.351 (dit "Sud-Africain")
Il a été découvert dans la région de Mandela Bay à l'est du Cap en Août 2020 (https://cov-lineages.org/global_report_B.1.351.html). Ce variant semble être très hautement transmissible car il est devenu la forme dominante du virus en Afrique du Sud dès le début Novembre 2020 avec aujourd'hui une prévalence de 90% (Tegally et al., MedRxiv 2020).

Séquence de la protéine S (spike) du variant Sud-Africain
La séquence est représentée ici (banque de données du National Center for Biotechnology Information; et Tegally et al., MedRxiv 2020) selon le code des acides aminés à une lettre. Les mutations par rapport à la séquence de la protéine S du virus "original" sont en rouge surilignées en jaune.
Il est maintenant présent dans au moins 32 pays dont la France (au moins 3 cas au 12 Janvier, selon le Conseil Scientifique COVID-19). Il comporte 21 mutations sur l'ensemble de son génome, dont 10 affectent la protéine S. Parmi ces 10 mutations, 3 se retrouvent dans le domaine de liaison de la protéine S au récepteur ACE2 (K417N, E484K, N501Y) (Lan et al., Nature 2020; Wang et al., Proc Natl Acad. Sci. 2020), dont 2 (E484K et N501Y) dans le motif de liaison au récepteur. Ce variant montre une vitesse de transmission accrue qui semble liée à la mutation N501Y. N501 interagit avec les résidus Y41 et K353 de ACE2 (voir sur ce site la page "Entrée du virus dans les cellules"). Dans un modèle de souris transgénique exprimant l'ACE2 humaine, cette mutation augmente l’affinité du virus pour ACE2. Les mutations K417N et E484K rares hors Afrique du Sud (<0,02%) aurait peu d’effet sur l’affinité avec ACE2 (Starr et al. Cell 2020). Le domaine de liaison au récepteur est la principale cible des anticorps neutralisants (Piccoli et al. Cell 2020). Les trois acides aminés K417, N501 et E484 établissent des liens avec des acides aminés des anticorps neutralisants. Des études ex-vivo (dans des cellules en culture) ont montré que la mutation E484K réduit l'effet neutralisant des anticorps monoclonaux et de divers anticorps polyclonaux spécifiques du SARS-CoV-2 isolés du sérum de personnes convalescentes (Weisblum et al., eLife 2020; Greaney et al., Cell Host & Microbes 2021). Selon ces études, ces mutations pourraient avoir un impact sur l’activité des vaccins ciblant la protéine S. De fait, une publication récente suggère que les vaccins à ARN Pfizer et Moderna pourraient être moins efficaces (50% au lieu de 95%) contre ce variant (Wu et al., BioRxiv 2021), mais des études supplémentaires sont nécessaires. Une étude non encore publiée portant sur environ 2 000 personnes menée à l’université du Witwatersrand à Johannesburg montrerait que le vaccin Oxford/AstraZeneca ne serait efficace qu’à 22 % contre les formes modérées de l'infection dues au variant Sud-Africain chez des personnes d’un âge moyen de 31 ans. Aucun résultat n’est en revanche disponible sur son efficacité contre les formes sévères liées à ce même variant (Journal International de Medecine 2021). L’équipe d’Oxford qui a co-développé ce vaccin avec AstraZeneka est déjà en train de travailler sur une version qui serait plus efficace contre ce variant et qui pourrait être prête à l’automne.
Le variant B.1.1.28.1 (dit "Brésilien")
Il a été découvert dans la ville de Manaus en Amazonie mi-Décembre 2020 (https://cov-lineages.org/global_report_P.1.html) est relativement proche du Sud-Africain. Après une large épidémie qui avait atteint son point culminant fin Avril 2020, les hospitalisations pour COVID-19 à Manaus sont restées stables et relativement faibles de Mai à Novembre, malgré l'assouplissement des mesures de confinement pendant cette période.

Séquence de la protéine S (spike) du variant Brésilien
La séquence est représentée ici (banque de données du National Center for Biotechnology Information; et Faria et al., Virological 2021) selon le code des acides aminés à une lettre. Les mutations par rapport à la séquence de la protéine S du virus "original" sont en rouge surilignées en jaune.
En octobre 2020, une étude sur les donneurs de sang indiquait que 76 % de la population avait été infectée par le SARS-CoV-2, non seulement à Manaus mais également dans d'autres endroits du bassin de l'Amazone. Le taux d'infection dans cette région serait donc supérieur au seuil théorique d'immunité de groupe (67 %). De fait, la brusque augmentation du nombre d'hospitalisations pour la COVID-19 à Manaus en Janvier 2021 (3431 du 1er au 19 Janvier 2021 contre 552 du 1er au 19 Décembre 2020) était inattendue et cette résurgence est très préoccupante (Préfecture de Manaus. Publication du 20 Janvier. COVID-19. http://www.manaus.am.gov.br/noticia/; Sabino et al., Lancet 2021). Elle pourrait être la conséquence du nouveau variant nommé B.1.1.28.1, P1. De plus, une femme de 37 ans (professionnelle de santé) résidant dans le nord-est du Brésil a présenté deux épisodes cliniques de COVID-19 en juin et octobre 2020, qui ont été confirmés par RT-PCR dans des échantillons prélevés à 116 jours d'intervalle (Resende et al. Virological 2021). Le séquençage du génome entier a révélé que les deux infections étaient causées, respectivement, par les deux lignées brésiliennes SARS-CoV-2 les plus répandues B.1.1.33 (primo-infection) et B.1.1.28 (réinfection). Ce variant B.1.1.28.1, P1 a également été détecté au Japon (après l'arrivée de 4 personnes provenant du Brésil) et aux US en Janvier 2021. Entre les 15 et 23 Décembre 2020, ce variant était détecté dans 42% des prélèvements PCR collectés par séquençage alors qu'il était absent des prélèvements réalisés entre Mars et Novembre 2020. Ces résultats indiquent une transmission intense et récente dans la région. Un séquençage systématique a montré qu'il comporte une constellation unique de 22 mutations incluant notamment K417T, E484K et N501Y, dont 2 (E484K et N501Y) dans le motif de liaison au récepteur (Faria et al., Virological 2021). Ces mutations qui sont également présentes dans le variant Sud-Africain. Des études ex-vivo (dans des cellules en culture) ont montré que la mutation E484K réduit l'effet neutralisant des anticorps monoclonaux et de divers anticorps polyclonaux spécifiiques du SARS-CoV-2 isolés de sérum de personnes convalescentes (Weisblum et al., eLife 2020; Greaney et al. Cell Host & Microbes 2021). Ces observations suggèrent que cette mutation entraîne un changement significatif d'antigénicité de la protéine S.
Le variant B.1.429 (dit "Sud-Californien")
Ce variant B.1.429 a été observé pour la première fois en juillet 2020 par des chercheurs du Cedars-Sinai Medical Center, en Californie, dans l'un des 1 230 échantillons de virus collectés à Los Angeles depuis le début de l'épidémie de COVID-19. Resté non détectable durant l'été 2020, il réapparut dans des prélèvements en Californie en Novembre 2020, date à partir de laquelle son incidence n'a cessé d'augmenter, passant de 36 % en Novembre à 50% en Janvier 2021.

Séquence de la protéine S (spike) du variant Californien
La séquence est représentée ici (banque de données du National Center for Biotechnology Information; et Zhang et al. JAMA 2021) selon le code des acides aminés à une lettre. Les mutations par rapport à la séquence de la protéine S du virus "original" sont en rouge surilignées en jaune.
B.1.429, est définie par 10 mutations ORF1a-T265I et -I4205V, ORF1b-P314L et D1183Y, S-S13I, -W152C, -L452R et -D614G, ORF3a-Q57H et N-T205I (Outbreak.info; Zhang et al. JAMA 2021; Zhang et al. MedRxiv2021). Parmi les 3 mutations distinctes dans la protéine S, la mutation L452R avait été précédemment détectée dans des variants non apparentés. Cette mutation se trouve dans le domaine de liaison au récepteur ACE2 et s'est avérée conférer une résistance à certains anticorps monoclonaux dirigés contre la protéine S dans des expériences in vitro (Li et al. Cell 2020). On sait que des mutations dans ce domaine peuvent également induire une résistance aux sérums polyclonaux comme on l'observe chez les patients convalescents ou chez ceux qui ont été vaccinés (Greaney et al. BioRxiv 2020). Ce variant semble plus transmissible et provoquer davantage de décès, mais des études supplémentaires seront nécessaires pour confirmer cela (Wadman, Science 2021). Dans un communiqué de presse conjoint de l'Université de Californie, San Francisco, du Département de santé publique de Californie et du Département de santé publique du comté de Santa Clara, le variant a également été détectée dans plusieurs comtés de Californie du Nord où sa fréquence est passée de 3 % à 25 % entre Novembre et Décembre 2020. Ce variant est maintenant détecté à des fréquences variables dans la plupart des États américains et quelques cas ont été détectés dans d'autres pays d'Amérique du Nord, d'Europe (dont la France), d'Asie et d'Australie (Outbreak.info).
Le variant B.1.526 (dit "New-Yorkais")
Ce variant avait été observé une première fois en Novembre 2020 dans les départements de microbiologie et de maladies infectieuses de l'Université Columbia à New-York, lors d'analyses de routine (Annavajhala et al. MedRxiv 2021; West et al. MedRxiv 2021). En fait, les cliniciens cherchaient à traquer rapidement l'apparition des variants B.1.1.7 (Britannique), B.1.351 (Sud-Africain) et B.1.1.28.1 P1 (Brésilien). Pour ce faire ils ont mis au point un test de RT-PCR adapté à la détection des mutations sur la protéine S, N501Y et E484K portées par ces variants. Les résultats ont montré une augmentation dramatique du nombre de cas de mutations E484K qui les a conduits à entreprendre un séquençage systématique du génome de ces variants. Et c'est lors de la comparaison phylogénétique de ces génomes viraux qu'est apparu le nouveau variant identifié sous le code B.1.526.

Séquence de la protéine S (spike) du variant New-Yorkais
La séquence est représentée ici (banque de données du National Center for Biotechnology Information; et Annavajhala et al. MedRxiv 2021) selon le code des acides aminés à une lettre. Les mutations par rapport à la séquence de la protéine S du virus "original" sont en rouge surilignées en jaune.
Outre la mutation E484K déjà trouvée chez les variants Britannique, Sud-Africain et Brésilien, ce variant comporte les mutations suivantes sur la protéine S: L5F, T95I, D253G, D614G, and A701V. Par la suite, il a été constaté une augmentation substantielle des cas positifs pour ce variant au fil du temps, de 1,3 % au début Novembre 2020 à 5,3 % à la mi-Janvier, et finalement à 12,3 % entre le 8 et le 15 Février 2021. Les autres mutations dans le génome du variant B.1.526 sont: N, P199L et M234I; NS3, P42L et Q57H; NS8, T11I; NSP2, T85I; NSP4, L438P; NSP6, del106-108; NSP12, P323L; et NSP13, Q88H. Ce variant semble s'étendre dans la région Nord de l'état de New-York et surtout dans tout le Nord-Est des Etats Unis. Le patient zéro de ce variant pourrait être un malade atteint du SIDA et qui avait contracté le SARS-CoV-2 en Août 2020 (Annavajhala et al. MedRxiv 2021). L'administration Biden surveille ce variant de très près.
Le variant B.1.617 (dit "Indien")
Le premier variant B.1.167 est apparu pour la première fois en Inde dans l'état de Maharashtra, le 5 octobre 2020. Il s'est ensuite rapidement répandu dans l'ensemble du pays au gré des rassemblements populaires de masse. A cet égard, le festival religieux hindou du Kumbh Mela censé apporter une chance de salut aux centaines de millions de pèlerins qui convergent vers le Gange, semble avoir surtout apporté le virus et la maladie. Ce festival pourrait avoir joué un rôle majeur dans l'épidémie bien qu'il soit impossible de déterminer son impact avec précision en absence de recherche des cas contacts. Mais, selon les responsables locaux, les chefs religieux et les médias, l'événement a probablement été l'une des sources majeures d'infection dès la mi-avril 2021.
Aujourd'hui (Mai 2021) le variant B.1.167 se décline en trois sous-variants, le B.1.617.1 (premier découvert), les B.1.617.2 et B.1.617.3 (découverts en décembre 2020). Outre l'Inde, les premiers cas de ces variants ont été détectés en février 2021 au Royaume-Uni, aux États-Unis et à Singapour, puis en avril au Canada, aux Iles Fidji et en Finlande. Ces variants sont aujourd'hui présents dans une douzaine de pays en Europe (dont la France avec environ 50 cas), en Afrique, en Australie, en Nouvelle Zélande, et en Corée du Sud.
En Inde, au pic de l'infection (mi-mai 2021), les variants B.1.167.1 à .3 ont produit plus de 400 000 cas de COVID-19 par jour avec plus de 4500 morts par jour. Au Royaume Uni, pays entretenant des relations culturelles, sociales et commerciales importantes avec l'Inde, le variant B.1.617.2 surpasse les deux autres variants B.1.167.1 et.3, et est aujourd'hui en train de monter en puissance. Les cas détectés liés à ce variant (séquençage du génome viral) sont passés de 1313 à 3424 au cours de la semaine du 19 mai, selon les données de Public Health England. Ce variant touche toujours principalement le nord-ouest de l'Angleterre et Londres, mais il existe des clusters dans tout le pays. Il pourrait être en passe de remplacer le variant B.1.1.7 (dit Britannique) et serait responsable de 50% des nouveaux cas de COVID-19 en étant 50% plus transmissible. Cependant cette poussée de B.1.617.2 n'entraîne pas d'augmentation significative du nombre total de cas au Royaume-Uni, qui reste bien inférieur à celui enregistré lors du dernier pic, en janvier 2021.

La séquence est représentée ici (banque de données du National Center for Biotechnology Information et European Centre for Disease Prevention and Control. "Threat Assessment Brief: Emergence of SARS-CoV-2 B.1.617 variants in India and situation in the EU/EEA – 11 May 2021" CDC; 2021) selon le code des acides aminés à une lettre. Les mutations par rapport à la séquence de la protéine S du virus "original" sont en rouge surilignées en jaune.
Les variants B.1.617.1 à .3 sont définis par au moins 8 mutations sur la protéine S : G142D, E154K, L452R, E484Q, D614G, P681R, Q1071H, H1101D. Quelques différences existent entre ces variants : le B.1.617.3 ne porte pas la mutation E484Q ; en revanche, le B.1.617.2 comporte la mutation T478K, absente des deux autres sous-variants. Neuf autres mutations caractérisent ces variants sur les régions ORF1ab, orf3a, orf6 et orf7a. La mutation L452R avait déjà été observée dans la protéine S du variant B.1.429 (dit Californien) voir plus haut. Cette mutation apparaît dans le domaine de liaison de la protéine virale S au récepteur ACE2 et s'est avérée conférer une résistance à certains anticorps monoclonaux dirigés contre la protéine S dans des expériences in vitro (Li et al. Cell 2020). La mutation en position 484 avait déjà été observée dans le variant B.1.1.28.P1 (dit Brésilien) et le variant B.1.351 (dit Sud-Africain), voir plus haut. La mutation en position 681 avait déjà été observée dans le variant B.1.1.7 (dit Britannique) et le variant B.1.351 (dit Sud-Africain), voir plus haut. Par ailleurs, il semble selon une étude récente que les trois mutations L452R, E484Q et P681R pourraient concourir à provoquer une transmissibilité accrue du virus (Cherian et al. BioRxiv 2021).
En théorie, la propagation accélérée du variant B.1.617.2 au Royaume-Uni, où plus de 50 % de la population a reçu au moins une dose de vaccin COVID-19, pourrait indiquer une capacité à échapper à la protection vaccinale. Mais il y a cependant peu de preuves que l'évasion vaccinale soit à l'origine de l'augmentation du nombre de cas. En effet, les données préliminaires recueillies mi-mai à Bolton et Blackburn, les deux villes les plus touchées par l'épidémie dans le nord-ouest de l'Angleterre, montrent que la plupart des personnes hospitalisées pour la COVID-19 causée par B.1.617.2 (confirmées par séquençage du génome viral) n'avaient pas été vaccinées. Cinq des 18 personnes hospitalisées dont le test PCR du variant B1.167.2 était positif avaient reçu une seule dose de vaccin, et une seule avait reçu les deux doses. D'autres données ont montré que les infections dues au variant B.1.617.2 dans le nord-ouest de l'Angleterre étaient initialement concentrées chez les adolescents qui ne sont pas systématiquement vaccinés. Bien que le variant se soit ensuite propagé aux trentenaires, aux quadragénaires et aux quinquagénaires - qui sont plus susceptibles d'avoir reçu les deux doses de vaccin – ces derniers n'ont pas connu de taux d'infection plus élevé. Certaines études in vitro indiquent cependant que les vaccins pourraient être moins efficaces contre le sous-type B.1.617.1. Les résultats d'expériences similaires avec B.1.617.2 n'ont pas encore été publiés, mais les données sur une population de personnes vaccinées ou non, ou ayant une ou deux doses (Bernal et al. Public Health England, 23 mai 2021) suggèrent que les vaccins Pfizer-BioNTech et Oxford-AstraZeneca sont efficaces (en termes de diminution des cas de formes graves et de décès) à 88 et 66%, respectivement, contre le variant B.1.617.2 après les deux doses.
Cet ensemble de données est donc rassurant et suggérent que la vaccination protège relativement bien de ce variant B.1.167.2. Il est enfin rassurant de constater qu'aucune mutation dans l'un des sous-types des variants B.1.617 n'est associée à une gravité accrue de la maladie.
Comment les variants sont-ils apparus ?
Etant donné le grand nombre de mutations par rapport au SARS-CoV-2 "original", une hypothèse permettant d'expliquer l'émergence de ces variants est qu'ils pourraient être apparus par évolution intra-hôte chez des personnes présentant une réplication virale prolongée sur plusieurs mois (Choi et al., New England Journal of Medicine 2020; Avanzato et al. Cell 2020) peut-être du fait d'une immunodéficience acquise ou pathologique. Voir à ce sujet le patient zéro du variant B.1.526 (New-Yorkais). Voir aussi par exemple sur ce site la page "Le syndrome Post-COVID-19 et malades au long cours". En effet, cette situation d'infection chronique favoriserait la prolifération des populations de virions et donc l'émergence de nouveaux variants. Il est à ce sujet intéressant de mentionner qu'en Afrique du Sud, le pays qui connaît la plus grande épidémie de VIH au monde, la possibilité d'une réplication virale prolongée et d'une évolution intra-hôte dans le contexte de l'infection par le VIH (un virus qui diminue très fortement l'activité du système immunitaire) est une vraie source de préoccupation. Cependant, les preuves que l'infection par le VIH soit associé à une réplication persistante du SARS-CoV-2 ne sont pas solides à ce jour. Il convient toutefois de réaliser que la diversité observée au sein des variants d'intérêt (Britannique, Sud-Africain et Brésilien) ne peut s'expliquer par une seule infection à long terme chez un seul individu. Si l'évolution par le biais d'infections à long terme peut être l'explication de l'évolution des variants, il faut aussi invoquer l'existence d'une chaîne de transmission du virus qui passe par plusieurs individus.
En revanche, l'évolution antigénique, même chez des individus non immunodéprimés, pourrait offrir une hypothèse alternative. En effet, plusieurs des épitopes de la protéine S (au sein du domaine de liaison au récepteur) semblent être sous une pression sélective élevée du fait que ces sites sont des cibles privilégiée des anticorps neutralisants. Le fait que plusieurs de ces mutations (K417N ou K417T, E484K, N501Y) aient été retrouvées dans les différents variants conforte cette hypothèse alternative selon laquelle l'émergence de ces mutations se serait faite en réponse à la pression de sélection représentée par les anticorps anti-S (Tegally et al. MedRxiv 2020).
Comment les variants sont-ils détectés ?
Deux méthodes: par RT-PCR ciblée ou par séquençage du génome.
Puisqu'on connait la position des mutations dans le génome des variants, on peut aisément mettre au point des tests PCR (voir sur ce site la page "COVID-19: les tests") qui sont directement ciblés sur ces positions. Rappelons que c'est de cette manière, et de façon fortuite, que le premier variant d'intérêt (B.1.1.7) a été découvert en Angleterre. Aujourd'hui ces tests PCR spécifiques des variants sont utilisés sur des personnes suspectées d'être infectées par un variant pour diverses raisons: retour de voyage, cas contact, situation géographique montrant l'existence d'un cluster, résurgence de l'infection détectée par analyses sur les eaux usées (voir sur ce site la page "Du SARS-CoV-2 dans les égouts"). Une fois qu'un variant a été détecté par PCR, il faut le confirmer par séquençage du génome entier.
Un réseau national de coordination sur les variants a été créée avec Santé Publique France, l'ANRS (Agence Nationale de Recherche sur le Sida et les maladies émergentes), les Centres Nationaux de Référence, les plateformes de séquençage haut débit, et les laboratoires de virologie. On espère d'ici quelques mois que la France pourra séquencer jusqu'à 4000 génome de virus SARS-CoV-2 et obtenir ainsi une image de la distribution des variants dans notre pays.
Conclusion
Après avoir initialement contenu la première vague de l'épidémie, de nombreux pays européens, américains et asiatiques ont connu une résurgence (deuxième vague) de la COVID-19 qui suggère qu'une grande partie de la population restait encore sensible au virus après la première vague épidémique. Si les nouveaux variants du SARS-CoV-2 ont une transmissibilité accrue par rapport aux variants circulants préexistants et s'ils sont associés à une résistance aux anticorps neutralisants, ils pourraient entraîner une résurgence de l'infection dans les lieux où ils circulent, comme ce fut le cas à Manaus. En France, au 27 Janvier, les variants d’intérêt Britannique, Sud-Africain et Brésilien sont encore minoritaires mais représentent tout de même 14% des cas détectés pour ce qui conerne le variant Britannique. C'est pour cette raison, qu'une surveillance rigoureuse aux plans génétique, immunologique, clinique et épidémiologique des nouveaux variants est indispensable. De plus, il est impératif de déterminer quelle est l'efficacité des vaccins anti-COVID-19 existants contre les divers variants manifestant des possibilités d'évasion immunitaire et notamment les 2 variants Sud-Africain et Brésilien. Le fait que ces deux variants comportent des mutations différentes mais également des mutations identiques (K417N/K417T, E484K, N501) situées dans le motif de liaison de la protéine S au récepteur ACE2 suggère que ces dernières pourraient avoir été acquises par avantage adaptatif et sélectif du virus plutôt que par le biais d'un effet fondateur (transmissions du virus à plusieurs personnes ou partir d'un unique individu porteur original du variant) ou par dérive génétique (mutations au hasard). Pour le moment, les variants Américains (Californien et New-Yorkais) restent discrets en Europe et en France.
Au plan théorique, il n'est pas certain que la résistance des variants aux anticorps neutralisants soit un élément suffisant pour réduire l'efficacité des vaccins, d'une part parce que les vaccins induisent des lymphocytes T-cytotoxiques qui, à côté des anticorps, ont un rôle protecteur majeur dans le contrôle de l'infection, et d'autre part parce que ces vaccins induisent un niveau très élevé d'anticorps neutralisants qui pourraient contrebalancer la résistance des variants aux anticorps. Autrement dit, les variants pourraient être moins sensibles aux anticorps neutralisants mais pas suffisamment pour entraîner un échec des vaccins (Moore et al., JAMA 2021). Il est en effet important de rappeler ici que les propriétés de résistance des variants aux anticorps naturels ou aux vaccins ont été obtenues sur les cellules en culture et non in vivo.
Compte tenu de l'essor de ces variants d'intérêt, plusieurs mesures devraient être prises.
Tout d'abord, les virus du SARS-CoV-2 prélevés sur des individus préalablement vaccinés mais néanmoins hospitalisés pour une COVID-19 (donc possiblement "réinfectés") doivent être immédiatement isolés et caractérisés. Cela serait un premier signe que des variants du virus "original" deviennent résistants à l'immunité induite par les vaccins actuels dirigés contre le virus "original".
Deuxièmement, les états doivent privilégier et maintenir un séquençage actif et un système de surveillance pour identifier rapidement ces variantes une fois qu'ils surgissent. Si le Royaume-Uni a été excellent à cet égard, une grande partie du reste du monde, et la France en particulier, ne l'a pas été. Nous avons vu ci-dessus qu'une coordination autour des centres de référence se met en place dans notre pays, mais il est peut-être un peu tard. Enfin, une coopération internationale sera essentielle.
Troisièmement, il serait indispensable de créer une banque centrale de prélèvement de sérums issus de personnes vaccinées contre le SARS-CoV-2. Ceci permettrait de tester les capacités des anticorps qu'ils contiennent à neutraliser les nouveaux variants au fur et à mesure qu'ils sont identifiés.
Quatrièmement, il est essentiel de réduire la propagation mondiale des nouveaux variants, en particulier les variant Sud-Africain et Brésilien. Bien que ces variants aient déjà commencé à circuler dans divers pays du monde, plus fréquemment ils seront réintroduits, plus ils risqueront de devenir de "super propagateurs".
Cinquièmement, les vaccins à ARNm et les vaccins adénoviraux à réplication défectueuse peuvent être adaptés assez rapidement pour prendre en compte les changements de séquence spécifiques (et déterminants) présents dans les nouveaux variants.
Sixièmement, comme ceux qui ont circulé tout au long de l'année 2020, les nouveaux variants se sont répandus par aérosolisation mais ne parcourent pas de longues distances. Porter les masques, et la distanciation physique peuvent empêcher leur propagation (Moore et al., JAMA 2021).
Enfin, d'autres variants d'intérêt devraient apparaître dans les prochains mois? Cela semble inéluctable. Wait and see, donc...
Date de dernière mise à jour : 29/07/2021
Commentaires
-
Comme d'habitude Patrick nous donne des informations claires sur un sujet complexe si mal décrit par nos média qui ne mettent en évidence que le "tragique" de la chose. Félicitation pour cet éclaircissement constructif.
Amitiés
Claude

Ajouter un commentaire