
Pas de manipulation
Pas d'évidence de manipulation humaine du SARS-CoV-2
Résumé tout public
L’interview récente du Professeur Luc Montagnier reprise par les médias, dans laquelle il suspecte une manipulation humaine d’un coronavirus - qui se serait traduite par la production du SARS-CoV-2 responsable de la pandémie actuelle de Covid-19 - n’a échappé à personne et a alimenté un grand nombre de débats et de forums sur internet.
Qu’en est-il exactement ?
Préambule
Les virus sont constitués d’une enveloppe ou d’une capside au sein de laquelle est empaqueté leur génome, ADN ou ARN. Entrer dans une cellule leur est indispensable pour se multiplier car ils ne possèdent pas la capacité de synthétiser des protéines. Ils comblent cette lacune en détournant à leur profit la machinerie cellulaire de synthèse des protéines. L’entrée du virus dans une cellule est donc un évènement déterminant pour sa survie. La première étape de ce processus est la formation d’un couple entre une protéine « S » de la surface du virus et une protéine « R » (récepteur) de la membrane des cellules. Pour le SARS-CoV-2, le couple S-R est tenu par la protéine virale appelée « Spike (S) » et la protéine membranaire appelée ACE2, le récepteur. C’est l’interaction entre la partie de la protéine S (qu’on appelle le domaine de liaison au récepteur) et la protéine ACE2 qui est à la base du tropisme d’espèce d’un virus et de son infectiosité. Si on souhaite rendre le SARS-CoV-2 particulièrement infectieux pour l’espèce humaine par des manipulations génétiques, il faut insérer dans la séquence de la protéine S, et en particulier dans le domaine de liaison au récepteur, des acides aminés qui permettront d’obtenir une interaction maximale avec ACE2.
L’annonce
Le 31 Janvier 2020 des chercheurs indiens publient un article dans lequel ils attirent l’attention sur la présence de 4 morceaux de protéines du virus du SIDA (HIV-1) dans la protéine « Spike (S) » du SARS-CoV-2. Ils affirment que ces morceaux de protéines du HIV-1 ne se retrouvent dans aucun séquence de coronavirus connue jusqu’ici, et que 3 d’entre eux sont localisés dans le domaine de liaison avec le récepteur ACE2. Pour eux, il ne peut s’agir d’une coïncidence.
La rumeur d’une manipulation d’un coronavirus est lancée…
Mise au point
Une analyse de divers articles publiés dans des revues scientifiques à comité de lecture montre les points suivants :
- Les morceaux de protéines du HIV-1 ne sont pas uniquement trouvés dans la protéine S du SARS-CoV-2, mais également dans la protéine S du coronavirus de chauve-souris et dans un grand nombre de protéines d’autres virus très divers (astrovirus, virus marins, papillomavirus bovin, etc.).
- Les 4 morceaux de protéines en question ne font pas partie du domaine de liaison de la protéine S avec son récepteur (ACE2).
- Les données accumulées depuis plus de 10 ans sur le SRAS-CoV (très proche du SARS-CoV-2) et en particulier sur le domaine de liaison de la protéine S avec son récepteur, suggèrent la « marche à suivre » pour construire en laboratoire un coronavirus dont la séquence lui confèrerait un pouvoir optimal d’infection de l’espèce humaine. Nul doute qu’un scientifique malveillant aurait choisi et facilement pu produire cette séquence s’il avait réellement manipulé le virus.
- Par contre la possibilité que le SARS-CoV-2 ait été étudié dans le laboratoire P4 de Wuhan, et ait pu « s'échapper » après une fausse manœuvre technique ou humaine, ne peut être ignorée.
Pour conclure, aucun élément objectif ne permet d'affirmer que le virus de Wuhan aurait été manipulé par des chercheurs.
Origine de cette affaire
Le 31 Janvier 2020 des chercheurs Indiens de l’Université de New Delhi publient dans BioRxiv (une plateforme de publication scientifique sans comité de lecture) un article intitulé : « Uncanny similarity of unique inserts in the 2019-nCoV spike protein to HIV-1 gp120 and Gag », soit explicitement en français : « Étrange présence dans la protéine de pointe du 2019-nCoV d’insertions similaires à celles présentes dans les protéines gp120 et Gag du VIH-1, virus responsable du SIDA ». Vous pouvez télécharger cet article ICI.
C’est cet article qui a lancé la polémique sur la possibilité d’une intervention humaine dans le design du SARS-CoV-2 et les dérives tous azimuts sur la théorie du complot chinois qui ont suivi, bien que les auteurs n’aient jamais utilisé ces termes de façon explicite.
Importance de la protéine S dans l’infectiosité du SARS-COV-2
Nous savons que pour survivre, tout virus doit trouver le moyen d’entrer dans les cellules bactériennes, végétales, animales ou humaines afin de s’y reproduire en utilisant la machinerie de synthèse des protéines des cellules elles-mêmes. Ce mécanisme d’entrée est basé sur la reconnaissance très spécifique par une des protéines de l’enveloppe virale d’une protéine de l’hôte qui va agir comme récepteur et permettre au virus de pénétrer la cellule. De nombreuses études ont permis d’établir que la sensibilité d’un hôte à une infection virale est principalement déterminée par l'affinité entre une protéine de l’enveloppe virale et une protéine récepteur localisée sur les membranes cellulaires de l'hôte (Wan et al. J Virol 2020). Dans le cas du SARS-COV-2, la protéine virale est la protéine S et le récepteur est la protéine humaine ACE2 (voir sur ce site ICI). Les analyses sur les protéines de la membrane des virus et sur leurs interactions avec les protéines récepteurs cellulaires sont donc d’une extrême importance car ce sont ces interactions qui sont à la base du tropisme d’espèce d’un virus et de son infectiosité. En d’autres termes et en bref, si on souhaite rendre un virus particulièrement infectieux pour l’espèce humaine par des manipulations génétiques, il faut insérer dans la séquence de la protéine virale les acides aminés qui permettront d’obtenir une interaction maximale avec le récepteur humain.
Article 1 : Pradhan et collaborateurs
En utilisant les bases de données du NCBI (National Center for Biotechnology Information) sur les séquences nucléotidiques et protéiniques et divers logiciels de comparaison de ces séquences (dont BLAST) ainsi qu’un logiciel de modélisation structurale de protéines en 3D, ces chercheurs ont réalisé une analyse du gène codant pour la protéine S du SARS-CoV-2 (dite « de pointe » impliquée dans la liaison du virus sur le récepteur à la surface des cellules humaines (voir sur ce site) qui a révélé les résultats suivants :
- Identification de 4 insertions IS1 (acides aminées 76 à 83), IS2 (aa 148-155), IS3 (aa 256-261), et IS4 (aa 681-688) dans la protéine S, insertions que, selon les auteurs, on ne retrouve dans aucun autre coronavirus séquencé jusqu'à présent.
- Ces 4 insertions sont également présentes dans les protéines Gp120 et Gag du HIV-1 (virus du SIDA).
- Selon les auteurs, cette étrange similitude n'est probablement pas fortuite.
- En outre, la modélisation 3D suggère qu'au moins 3 de ces insertions sont des composants clés du site de liaison du virus au récepteur. En fait la structure 3D établie dans ce travail n’est pas confirmée par la structure 3D du complexe protéine virale S et récepteur ACE2 obtenue par cryo-microscopie électronique et cristallographie aux rayons X (Yan et al. Science 2020 ; Lan et al. Nature 2020).
Les auteurs concluent que pris dans leur ensemble ces résultats suggèrent une évolution non conventionnelle du SARS-COV-2 qui justifie une étude plus approfondie sur l'évolution, la pathogénèse et le diagnostic de ce virus.
A noter que cet article a été retiré très rapidement de la plateforme BioRxiv par les auteurs, après avoir enregistré de multiples critiques au plan strictement scientifique (et non sous une quelconque pression des autorités chinoises comme affirmé par certains) comme nous allons le voir dans ce qui suit.
Article 2 : Zhang et collaborateurs
Cet article est intitulé : « Protein Structure and Sequence Reanalysis of 2019-nCoV Genome Refutes Snakes as Its Intermediate Host and the Unique Similarity between Its Spike Protein Insertions and HIV‑1», soit explicitement en français : « Une réanalyse de la séquence et de la structure de la protéine S du SARS-COV-2 réfute le serpent comme espèce-hôte intermédiaire et les similitudes entre la protéine S et les protéines du HIV-1 » a été publié le 22 Mars 2020 dans The Journal of Proteome Research. A télécharger ICI.
Ces chercheurs ont utilisé les bases de données du NCBI (comme Pradhan et al.) sur les séquences nucléotidiques et protéiques et d’autres approches expérimentales sur les comparaisons de ces séquences, ainsi qu’une modélisation en 3D de l’interaction entre la protéine S du virus et le récepteur humain, la protéine ACE2 (voir sur ce site).
Les résultats obtenus ont été les suivants :
- Il existe effectivement 4 insertions (IS1, IS2, IS3 et IS4) de 6 à 8 acides aminés dans la protéine S que l’on ne retrouve ni dans la protéine S du SRAS-CoV, ni du MERS-CoV.
- Contrairement à ce qui a été publié par Pradhan et al., les 4 insertions présentent dans la séquence de la protéine S se retrouvent dans des protéines d’autres virus, et notamment dans le coronavirus de chauve-souris RaTG13, voir la Table ci-dessous. Ceci suggère que ces insertions dans la protéine S de SARS-COV-2 ont été directement ou indirectement (à travers l’espèce intermédiaire) héritées du coronavirus de chauve-souris. Ce résultat est en accord avec le fait que le réservoir principal de ce virus est la chauve-souris.
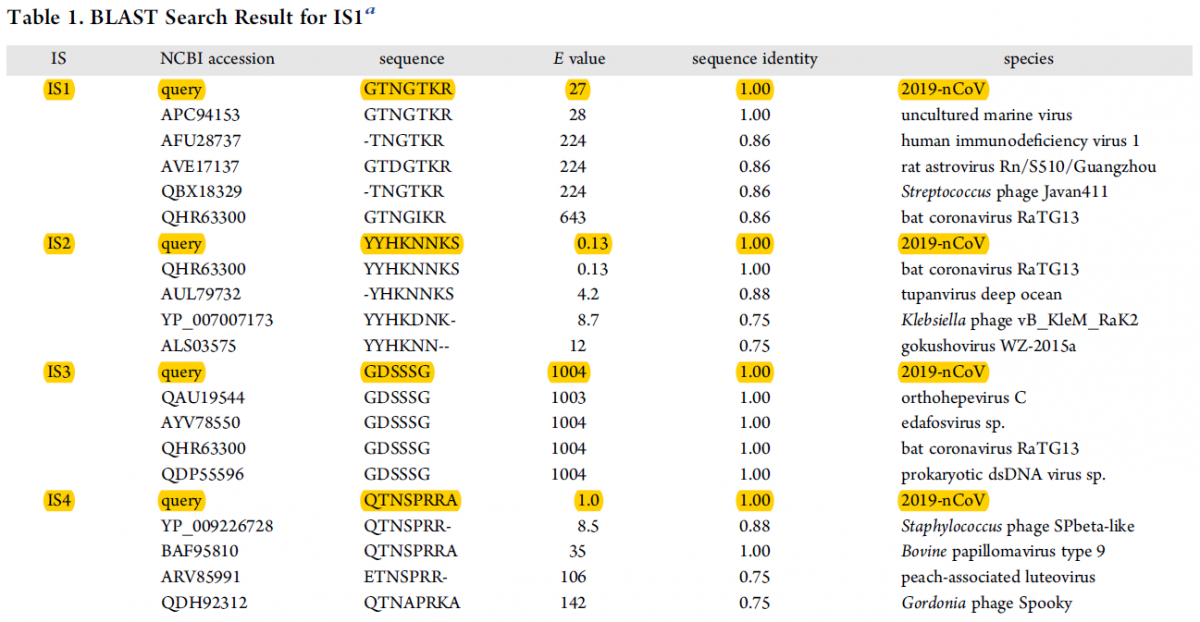
Table : Analyse comparative par BLAST (Basic Local Alignment Research Tool) des séquences des insertions IS1 à IS4 avec les données du NCBI.
BLAST est un outil de recherche heuristique utilisé en bio-informatique afin de trouver des régions similaires entre deux ou plusieurs séquences de nucléotides ou d'acides aminés, et de réaliser un alignement de ces régions homologues pour déterminer leur degré de proximité. La valeur E (valeur attendue) est un nombre qui décrit combien de fois on s'attend à ce qu'une séquence correspondante de cette taille soit trouvée par hasard dans une base de données. Plus la valeur E est faible, plus la concordance est significative. Colonne 1 : les insertions IS1 à IS4. Colonne 2 : numéro d’accession à la séquence considérée dans le base de NCBI. Colonne 3 : séquence en acides aminés avec le code à une lettre. Colonne 4 : valeur du paramètre E (voir ci-dessus). Colonne 5 : Sequence identity : identité de séquence : la valeur de 1 signifie une identité complète ; les valeurs inférieures traduisent une identité incomplète. Colonne 6 : les différentes espèces de virus qui comportent les séquences IS1 à IS4, soient en totale identité, soit en identité partielle (Selon Zhang et al. The Journal of Proteome Research 2020)
- La structure 3D établie dans ce travail (voir figure ci-dessous) est conforme à celle obtenue par cryo-microscopie électronique et cristallographie aux rayons X (Yan et al. Science 2020 ; Lan et al. Nature 2020). Contrairement à ce qui a été publié par Pradhan et al., aucune de ces 4 insertions se trouve dans le domaine de liaison (RBD) de virus au récepteur qui a été localisé entre les acides aminés 319 et 529 (Wan et al. J Virol 2020 ; Yan et al. Science 2020 ; Lan et al. Nature 2020) (voir sur ce site ici).

Figure. Structure modélisée en 3D du complexe entre la protéine S (skipe) du virus et la protéine ACE2 récepteur humaine.
La protéine ACE2 est représentée à gauche (en jaune) et la protéine S est à droite sous forme d’un trimère (en vert, bleu et magenta) comme elle se présente sur l’enveloppe du virus. Les séquences IS1 à IS4 sont identifiées par les flèches noires. L’interface entre spike et ACE2 est signalé par une flèche. (Selon Zhang et al. The Journal of Proteome Research 2020).
Article 3 : Wan et collaborateurs
Cet article intitulé « Receptor Recognition by the Novel Coronavirus from Wuhan: an Analysis Based on Decade-Long Structural Studies of SARS Coronavirus», soit explicitement en français : « Récepteur du nouveau coronavirus de Wuhan : analyses basées sur dix années d’études sur le coronavirus du SRAS » a été publié dans The Journal of Virology 2020 (à télécharger ICI). Il examine en détail les interactions entre le domaine de liaison de la protéine virale S avec son récepteur humain, la protéine ACE2.
Les résultats sont les suivants :
- La séquence du domaine de liaison au récepteur de la protéine S du SARS-COV-2, y compris son motif de liaison au récepteur (RBM) qui contacte directement l'ACE2, est similaire à celle du SRAS-CoV, ce qui suggère fortement que le SARS-COV-2 utilise l'ACE2 comme récepteur (voir sur ce site ici).
- Deuxièmement, plusieurs acides aminés présents dans le motif de liaison au récepteur (RBM) du SARS-COV-2 et notamment la glutamine 493 permettent des interactions favorables avec l'ACE2 humain.
- Troisièmement, plusieurs autres acides aminés du RBM et en particulier l’asparagine 501, sont compatibles mais pas idéaux pour permettre la meilleure interaction avec le récepteur humain ACE2, suggérant que le SARS-COV-2 a acquis une capacité de transmission humaine non optimale.
Et c’est bien là le point essentiel qui va totalement à l’encontre d’une manipulation malveillante du coronavirus de Wuhan. Les auteurs concluent : il est alarmant (c’est exactement le terme qu’ils utilisent) de réaliser que selon les prédictions acquises depuis une dizaine d’années, une seule mutation se traduisant par le remplacement de l’asparagine en position 501 par une thréonine pourrait significativement renforcer l'affinité de liaison entre le SARS-COV-2 et l'ACE2 humaine et donc son potentiel infectieux. Ainsi, l'évolution de la séquence de la protéine S du SARS-COV-2 chez les patients doit être surveillée de près pour détecter l'apparition de nouvelles mutations à la position 501 et dans une moindre mesure, également à la position 494 (sérine).
Article 4 : Andersen et collaborateurs
Dans un article publié le 17 Mars 2020 par Andersen et al., intitulé « The proximal origin of SARS-CoV-2 » (en français : Origine probable du SARS-COV-2) dans la revue Nature Medicine, les auteurs envisagent plusieurs scénarios qui peuvent expliquer de manière plausible l'origine du SRAS-CoV-2 :
- Sélection naturelle chez un animal hôte (chauve-souris, puis passage à un intermédiaire, pangolin ? ) avant le transfert à l’homme. C’est l’hypothèse couramment admise aujourd’hui, même si l’espèce intermédiaire n’est pas encore clairement identifiée.
- Sélection naturelle chez l'homme à la suite d'un transfert de zoonose. Dans cette hypothèse, il est possible qu'un progéniteur du SRAS-CoV-2 ait pu passer chez les humains à partir d’un animal (chauve-souris, autre ?) et ait acquis les caractéristiques génomiques lui permettant de s'adapter à la transmission interhumaine. Une telle infection asymptomatique se serait produite entre Novembre et Décembre 2019. Une fois acquises, ces adaptations auraient pu permettre la pandémie que nous connaissons aujourd’hui.
- La recherche fondamentale en virologie se fait en laboratoires de confinement (P2, P3 ou P4) dans lesquels les virus sont cultivés sur des cellules animales ou humaines. Depuis de nombreuses années des cas documentés d'évasions de laboratoire de divers virus et du SRAS-CoV en particulier ont été documentées. La possibilité que le SARS-COV-2 ait été cultivé et étudié dans le laboratoire P4 de Wuhan, et ait malencontreusement pu « s'échapper » du laboratoire après une fausse manœuvre technique ou humaine, ne peut être ignorée.
Conclusion
Il n’y a pas eu de manipulation humaine à l’origine du SARS-COV-2 responsable de la pandémie de Covid-19.
La confrontation entre les articles 1 et 2 montre que les résultats de Prahan et al. sont incorrects sur deux points majeurs :
- au niveau des études comparatives des séquences d’insertion entre le SARS-COV-2 et d’autres virus. Ces auteurs n’ont pas vu que la protéine S du coronavirus de la chauve-souris comportait les mêmes insertions IS1, IS2, IS3 ou une insertion quasi identique (IS4); ils n’ont pas vu non plus que ces 4 insertions se retrouvent dans une multitude de virus totalement différents du HIV-1 et des coronavirus.
- au niveau du positionnement des insertions I1 à I4 dans le domaine de liaison du virus à son hôte. Ces auteurs ont incorrectement placé ces insertions dans le domaine de liaison au récepteur, ce qui n’est pas le cas, leur modèle 3D étant en total désaccord avec celui obtenu par cryo-microscopie électronique et cristallographie aux rayons X, par Yan et al. Science 2020 et Lan et al. Nature 2020.
Le fait que les 4 insertions se retrouvent à l’identique (IS1, IS2 et IS3) ou très proche (IS4) dans le coronavirus de la chauve-souris suggère que ce virus nous a été transmis par cette espèce, éventuellement via un hôte intermédiaire dont la nature reste à déterminer, plutôt que via des manipulations réalisées en laboratoire.
L’article 3 apporte des éléments déterminants dans la mesure où il montre que l’interaction entre le virus et son récepteur, la protéine ACE2, est loin d’être optimal. En fait, les données accumulées depuis plus de 10 ans sur les coronavirus humains en particulier sur le SRAS-COV (très proche du SARS-COV-2) suggèrent la « marche à suivre » pour construire en laboratoire un coronavirus avec un pouvoir optimal d’infection de l’espèce humaine. Nul doute qu’un scientifique malveillant aurait choisi la séquence la plus adaptée avec notamment l’acide aminé thréonine en position 501 de la protéine S.
Selon les arguments développés dans les articles 3 et 4, il est donc improbable que le SARS-CoV-2 soit apparu à la suite de manipulations en laboratoire d'un coronavirus apparenté au SRAS-CoV.
Alors, pourquoi les insertions IS1 à IS4 apparaissent-elles dans le HIV-1 (virus du SIDA) et dans une multitude d'autres virus (Voir la Table présentée ci-dessus) ? La raison première est que ces insertions sont relativement courtes. Plus un fragment de séquence nucléotidique (ou protéique) est court et plus la chance de le trouver dans divers gènes ou génomes viraux est élevée. C'est implicitement ce qu'indiquent les résultats présentés dans le Table ci-dessus et notamment les valeurs du paramètre E. Je rappelle ici que plus la valeur E est faible, plus la concordance est significative. Exemple, la valeur de E de 0.13 pour la séquence de IS2 trouvée dans le coronavirus de la chauve-souris (RaTG13). Au contraire plus la valeur de E est forte (supérieure à 1) et plus la chance de trouver la séquence en question dans d'autres organismes (virus notamment) est forte. Ainsi la présence des séquences IS1 à IS4 dans le génome du HIV-1 (virus du SIDA) n'est que fortuite. Enfin, il faut bien voir que les insertions IS1 à IS4 ne sont pas 100% identiques à celles présentes dans la protéine S de SARS-COV-2. Exemple de IS1: 86% de similitude avec le HIV-1, et bien moins pour d'autres.
Date de dernière mise à jour : 21/04/2021
Commentaires
-
Très bien Patrick pour ce résumé "grand public" qui permet, avec des mots simples de se faire une opinion sur les avancées de la virologie, mais également sur la "pertinence" des informations journalistiques !! Comme tu sais, la science, ce n'est pas du baratin, mais ce sont des faits contrôlés et reproductibles par d'autres que par les auteurs des articles.
A te lire de nouveau, bien amicalement,
Claude -
article surement tres instructif sur l'etat des connaissances des chercheurs experts en virologie .Je suis preneuse d'une version simplifiée egalement . Victoire Chateau
-
article surement tres instructif sur l'etat des connaissances des chercheurs experts en virologie .Je suis preneuse d'une version simplifiée egalement . Victoire Chateau
-
Bonjour Mr Maurel,
Enfin une explication et surtout une information documentée qui permet d'y voir plus clair à travers cette information et désinformation dont on est abreuvées tous les jours. On a du mal à s'y retrouver si ce n'est en faisant l'effort de chercher, de recouper, de dépasser tout ce qui relève de la désinformation officielle, de garder la raison, et de ne rien prendre pour argent comptant sans analyse critique.
Sur un autre sujet, après avoir "déguster" le roman la vois des autres je suis en prise actuellement avec Khonsou et le Papillon que mon Beau Frère Jean claude m'a signalé et je dois le dire, je trouve toujours le même plaisir à vous lire.
Pierre BORD
Encore Merci pour vos précision -
Merci Patrick pour cette analyse détaillée et scientifiquement très étayée. J'aurais une suggestion, c'est que tu en fasses une version "grand public" de manière à ce que nous puissions, si tu en es d'accord, évidemment, à la diffuser parmi nos amis non scientifiques qui doivent "gober" les informations journalistiques souvent fantaisistes....
Bien amicalement,
Claude -
Merci Patrick pour cette page qui a le mérite d'être argumentée.
Ce qui n'est pas le cas de ce que l'on peut entendre au quotidien.
Bonne continuation.
Jc -
MERCI Patrick pour ces infos qui s'appuient sur des données concrètes. C'est essentiel pour ne pas basculer dans la paranoïa.
Bon confinement
Sabine





Ajouter un commentaire