Dans un article publié en Février 2017 dans la revue Reprod. Biomed. Online, le Dr John Zhang et son équipe décrivent les diverses étapes et les résultats des manipulations qui ont conduit à la naissance du premier “enfant aux trois parents”, le 6 avril 2016.
J’ai parlé de la thérapie par remplacement mitochondrial, ou plus de façon plus imagée, de “l’enfant aux trois parents” dans un dossier récent sur ce site (s’y référer pour la suite de cet article, si nécessaire).
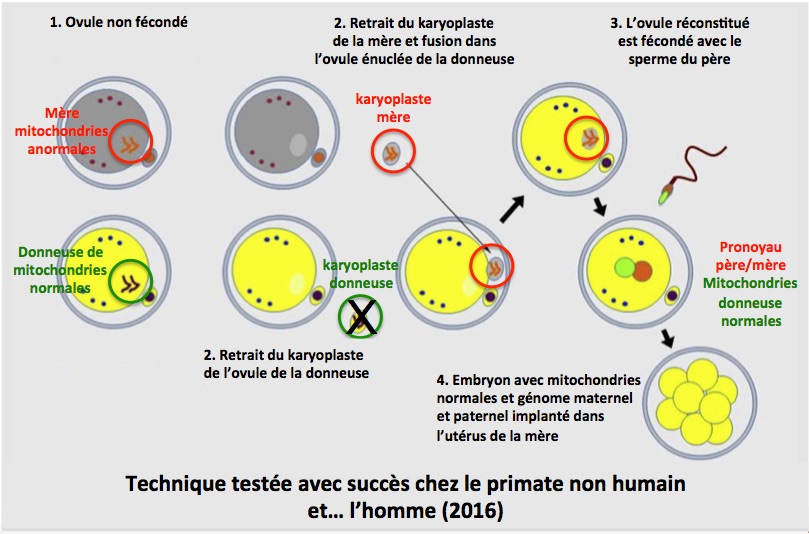
Cette thérapie a été entreprise car la mère était porteuse dans son ADN mitochondrial d’une mutation (8993T>G, responsable du syndrome de Leigh) qui compromettait grandement la vie de l’enfant (deux enfants décédés précédemment dans cette famille). Pour des raisons religieuses, la famille ayant refusé la destruction d’un embryon, la technique du transfert du karyoplaste (struture comprenant les chromosomes et le fuseau mitotique) dans l’ovocyte non fécondé a été utilisée. Très brièvement, voici les diverses étapes de cette technique (Figure):
1. Des ovocytes non fécondés de la future mère (porteuse de mitochondries anormales) et de la donneuse de mitochondries normales sont recueillis.
2. Le karyoplaste de la donneuse est retiré et éliminé. Le karyoplaste de la mère est prélevé et introduit dans l’ovocyte énucléé de la donneuse.
3. L’ovocyte ainsi réconstitué est fécondé avec le sperme du père.
4. L’embryon qui contient maintenant les mitochondries normales de la donneuse et les chromosomes de la mère et du père est implanté dans l’utérus de la mère.
Voici un résumé des données expérimentales publiées par John Zhang et son équipe :
- L'ADN de l’embryon sélectionné a été analysé par hybridation génomique comparative pour vérifier que son génome était normal (karyotype 46, XY). Par exemple, le nombre de copies (de gènes) peut augmenter en cas de duplication ou diminuer en cas de délétion, ou présenter des anomalies en cas d’aberrations chromosomiques.
- A la naissance, qui s’est passée normalement (après une grossesse sans histoire), l’enfant pesait 3,18 kg et mesurait 51,5 cm.
- Les séquences des ADNmt de la mère, de la donneuse de mitochondries et de l’embryon ont été comparées. Cette analyse a confirmé la présence de la mutation 8993T>G chez la mère. Ce résultat était attendu puisque deux précédents enfants étaient décédés (à 8 mois et 6 ans) de la madie de Leigh. Les niveaux d’hétéroplasmie chez la mère étaient de 23.27% dans les follicules pileux, 24.50% dans le sang et 33.65% dans l’urine. La mutation 8993T>G était évidemment absente chez la donneuse de mitochondries.
- Au jour 2 post-natal, le taux moyen de transmission de la mutation chez l’enfant était de 5,1±1,1% et le niveau moyen d’hétéroplasmie était de 5,7% mais variable selon les tissus, indétectable dans le placenta et le sang de cordon, 2.36% dans l’urine, 3.52% dans l’épithelium buccal, 5.59% dans les follicules pileux (cheveux), et 9.23% dans le prépuce.
Comme je l’ai déjà discuté sur ce site, cette méthodologie, pour aussi brillante qu’elle soit, pose des questions éthiques et biologiques. Je me limiterai ici à évoquer les questions biologiques. L’un des problèmes inhérants à cette méthodologie est le fait qu’il n’est pas possible de prélever le karyoplaste de l’ovocyte de la mère sans entraîner une petite fraction du cytoplasme, et donc, des mitochondries dont l’ADN contient la mutation fatale (ici la mutation 8993T>G qui conduit au syndrome de Leigh).
Or, les génomes nucléaire et mitochondrial ont évolué de concert depuis des centaines de millions d’années, et de cette co-évolution ont résulté des interactions fonctionnelles entre les protéines issues de gènes nucléaires et les protéines issues des gènes mitochondriaux. La question majeure est : ces interactions resteront-elles les mêmes après transfert quand les gènes nucléaires et mitochondriaux proviendront de deux individus différents ? Deux équipes ont récemment montré que cette possibilité existe au moins chez l’animal (Reinhardt 2013; Yamada 2016). C’est d’ailleurs la raison pour laquelle la “Human Fertilization and Embryology Authority” (HFEA) au Royaume uni et la FDA aux Etats Unis ont demandé une suspension des autorisations sur cette technologie jusqu’à ce que des études complémentaires soient réalisées sur ce problème.
Pour la mutation 8993T>G des études précédentes, réalisées à partir de 178 individus (Dahl 2000), ont montré que la probabilité de survenue de la maladie nécessite un taux de mutation de 60-70%, et que ni le taux de cette mutation ni le taux d’hétéroplasmie ne varient en fonction du temps. Selon John Zhang et son équipe, le fait que les taux de mutation et d’hétéroplasmie chez l’enfant soient inféreurs à 10% suggère qu’il ne sera pas atteint par la maladie.
(18 Avril 2017)

{kind=link}
{kind=link}