
Mucoviscidose
Kaftrio
La trithérapie qui change la vie

Une maladie Européenne
La mucoviscidose touche plus de 90 000 personnes au niveau mondial. En France on dénombrerait aujourd’hui plus de 7 000 malades. Des études récentes sur de l’ADN ancien, issu de restes humains prélevés sur divers sites archéologiques, montrent que cette maladie serait apparue par le biais d'une mutation fondatrice (F508del, voir plus loin) durant l’âge de bronze, il y aurait environ 5000 ans, dans le sud-ouest de l’Europe, sur les côtes de l’océan Atlantique et peut-être dans des régions de France ou du Portugal (Farrell 2018). Elle se serait ensuite étendue au reste de l’Europe centrale et orientale par les échanges entretenus par les peuples de culture campaniforme. Hormis dans les populations issues de croisements avec des ascendants Européens, cette maladie est peu présente dans le reste du monde.
En génétique, une mutation fondatrice est une mutation qui apparaît dans l’ADN d’un (ou plusieurs) individu et qui, à la suite de transmission à sa descendance, va créer une population particulière aux caractéristiques génotypique et phénotypique spécifiques. En génétique des populations, l’effet fondateur résulte de la perte de variations génétiques au sein d’une population particulière, établie à partir d’un petit nombre d’individus du fait par exemple d’un isolement géographique ou communautariste.
Une maladie génétique
C’est une maladie génétique récessive (c’est à dire qu’elle ne se déclare que si les deux parents du malade sont porteurs) due à une mutation dans la séquence du gène CFTR (Cystic Fibrosis Transmembrane Regulator, ou en Français, Régulateur de la Conductance Transmembranaire de la Mucoviscidose) porté par le chromosome 7. Ce gène code pour la protéine du même nom. La protéine CFTR est un canal anionique qui tapissent les cellules de plusieurs organes et permet le passage d’ions chlorure et bicarbonate de l’intérieur vers l’extérieur, assurant ainsi la régulation de la production de mucus, de sueur et d'autres fluides. De plus cette protéine joue un rôle important dans les macrophages (cellules participant à l’immunité en phagocytant les débris cellulaires et les agents pathogènes comme par exemple les bactéries) au niveau de la capture et de la destruction des bactéries qui y sont internalisées.
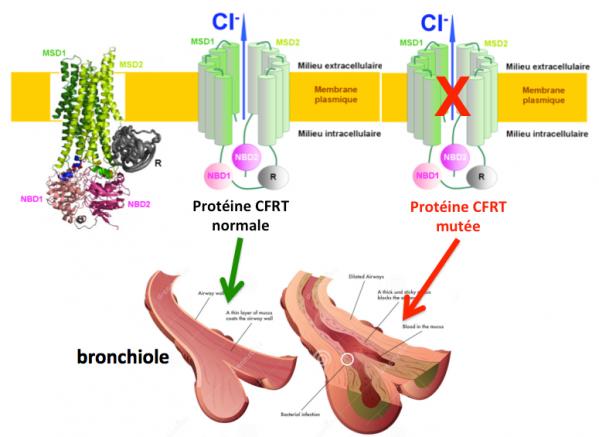
La protéine CFTR
Partie haute à gauche, la structure de la protéine obtenue par diffraction des rayons X, est présentée. Au centre et à droite, la structure de la protéine est présentée de façon schématique. Dans sa configuration normale, la protéine forme un canal qui permet le passage des ions chlorure (Cl-) et autres. Lorsque la protéine est mutée le canal ionique ne fonctionne plus. C’est à dire que le transport des ions chlorure ou bicarbonate est réduit ou bloqué et il en résulte des sécrétions épaisses qui obstruent les bronchioles pulmonaires ou les ductules pancréatiques, conduisant à la pathologie (partie basse droite de la figure).
Chez plus de 80% des malades dans la population Européenne (82% en France), la mucoviscidose résulte d’une mutation appelée F508del sur le gène CFTR ; cela signifie que l’acide aminé phénylalanine (F) qui devrait se trouver en position 508 de la séquence de la protéine est absent (del pour délétion), précisément du fait de la mutation. L’absence de cet acide aminé confère à la protéine CFTR mutée un mauvais repliement (une structure en 3D anormale) qui a deux conséquences majeures : i) elle est dégradée avant son transport sur la membrane des cellules ; ii) elle est inactive (le canal anionique reste fermé). Ces deux dysfonctionnements se conjuguent. La fonction biologique de la protéine est ainsi perdue, d’où la maladie. Plusieurs organes sont touchés: poumons, pancréas, intestin, foie, mais c’est l’atteinte broncho pulmonaire qui est la principale cause de morbidité et de mortalité de cette maladie. La mutation F508del, responsable de plus de 80% des cas de mucoviscidose, est décrite ci-dessous.
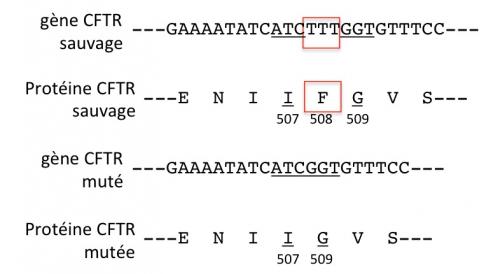
La mutation F508del
Dans le gène CFTR muté (troisème ligne à partir du haut), le triplet TTT encadré en rouge, présent sur le gène sauvage (normal, première ligne) est absent (délétion). Ce triplet code pour l'acide aminé phénylalanine (F) en position 508 sur la protéine sauvage (normale, deuxième ligne). Dans la protéine normale, l'acide aminé F est entouré des acides aminés isoleucine (I, posittion 507) et glycine (G, position 509). Dans la protéine CFTR mutée (ligne du bas) l'acide aminé phénylalanine est absent. Ce manque d'un seul acide aminé sur la protéine lui confère des propriétés différentes de sa forme sauvage (normale). D'où la maladie. Les traits en pointillés de part et d'autre des lettres figurent le reste de la séquence du gène et de la protéine, non montré ici.
Outre la mutation F508del, plus de 2000 autres mutations du gène CFTR ont été identifiés à ce jour et environ 700 d'entre-elles peuvent être à l'origine de la maladie. De nombreuses combinaisons de mutations peuvent ainsi se retrouver chez les malades. Dans notre pays, 82% des malades sont porteurs d’au moins une mutation F508del, soit parce qu’ils sont homozygotes pour cette mutation (c’est à dire que les deux parents en sont porteurs), soit parce qu’ils sont hétérozygotes (un des parents est porteur de F508del et l’autre parent est porteur d’une autre mutation différente). Alors que 1 Européen sur 25 à 40 est porteur de la mutation F508del, ce chiffre passe à 1 sur 17 000 pour les populations Africaines, 1 sur 11 000 sur les populations Amérindiennes et 1 sur 31 000 sur les population Asiatiques.
Caractéristiques cliniques
Les caractéristiques cliniques de la mucoviscidose résultent d'une fonction réduite ou nulle de la protéine CFTR. Cette protéine est présente dans les poumons, le pancréas, le foie, et le tractus gastro-intestinal. Ainsi par exemple dans le poumon la protéine CFTR est située dans la lumière des bronches et bronchioles ; dans le pancréas elle est située dans la lumière du canal pancréatique. Au niveau des poumons, l’absence ou l’inactivité de la protéine a pour conséquences un épaississement du mucus endo bronchique conduisant à une accumulation de sécrétions et à des infections (notamment par Pseudomonas aeruginosa), d’où une inflammation chronique conduisant à de graves problèmes respiratoires. Au niveau du pancréas, l’absence de la protéine conduit à une émission réduite des sucs pancréatiques conduisant à des troubles digestifs sévères. D’autres manifestations sont la cirrhose biliaire, l'absence de spermatozoïdes, et la sinusite chronique (Ong 2023).
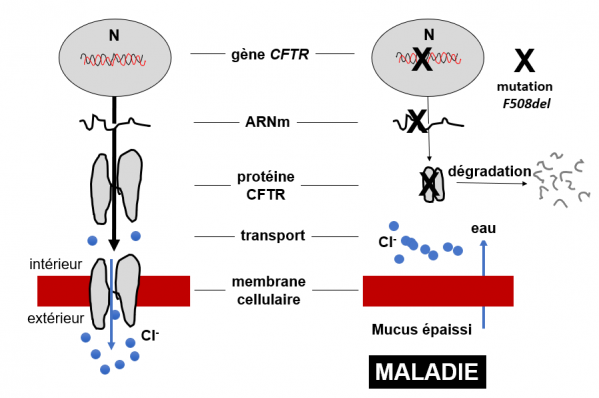
La mucoviscidose
Le schéma de gauche décrit la succession des étapes qui conduit du gène CFTR à la protéine CFTR et à sa fonction. Le gène CFTR est transcrit dans le noyau des cellules (N) en ARN messager (ARNm) qui passe dans le cytoplasme ou il est traduit en protéine CFTR par les ribosomes. La protéine est ensuite transportée par un système spécialisé (incluant le réticulum endoplasmique et l'appareil de Golgi) vers la membrane cellulaire (rectangle marron) au sein de laquelle elle va s'inserrer. Dans la membrane, la protéine forme un canal qui permet aux anions chlorures (Cl-) et bicarbonates de passer de l'intérieur vers l'extérieur des cellules par un processus énergétique généré grâce à l'ATP. Le schéma de droite montre la situation chez les malades porteurs de la mutation F508del (ou d'autres mutations) matérialisée par la lettre X. Cette mutation présente initialement sur le gène CFTR se retrouve sur l'ARNm et sur la protéine. Dans cette version mutée, la protéine est non seulement inactive et rapidement dégradée à l'intérieur de la cellules mais elle n'est pas prise en charge par le système de transport vers la membrane. Les anions (Cl-) et bicarbonates s'accumulent dans la cellule, ce qui conduit à une entrée d'eau par équilibre osmotique, et par conséquence à une concentration et un épaississement du mucus extracellulaire. C'est ce phénomèe qui conduit à la maladie.
Les traitements de la mucoviscidose
Pendant de nombreuses années, la mucoviscidose a représenté une maladie limitant sévèrement l'espérance de vie des malades. Alors qu’en 1950 l’espérance de vie d’un nouveau-né atteint ne dépassait pas une année, celle-ci est passée à 5 ans dans les années 1960, puis graduellement à 40-50 ans jusqu’aux années 2015, grâce aux progrès de la recherche et à l'amélioration de la prise en charge de la maladie. Jusqu’en 2019, l’arsenal des traitements symptomatiques de cette maladie comprenait les antibiotiques, les anti-inflammatoires, les composés mucolytiques, une alimentation riche en aliments à haute teneur calorique et en protéines et, en dernier recours, la transplantation pulmonaire.
Après l’identification du gène CFTR, de nombreux efforts se sont concentrés sur les approches de thérapie génique, malheureusement sans grand succès pour le moment. Cependant, des études sur la correction des mutations génétiques par la technique de l’édition génomique sont en cours actuellement.
La trithérapie de "modulateurs" : Kaftrio
Une autre stratégie thérapeutique a été mise en place par les chercheurs dans la décennie 2010 afin de corriger les défaillances fonctionnelles de la protéine CFTR mutée. Le processus de découverte de cette trithérapie a nécessité des efforts gigantesques. En effet, dans cette approche, on utilise un modèle cellulaire pulmonaire (à partir de prélèvements réalisés chez des malades) que l’on soumet in vitro à diverses molécules issues de banques de plusieurs millions de produits chimiques de synthèse. Ensuite on évalue l’effet de chacun de ces produits sur le transport des ions chlorures et/ou bicarbonates dans ce modèle cellulaire.
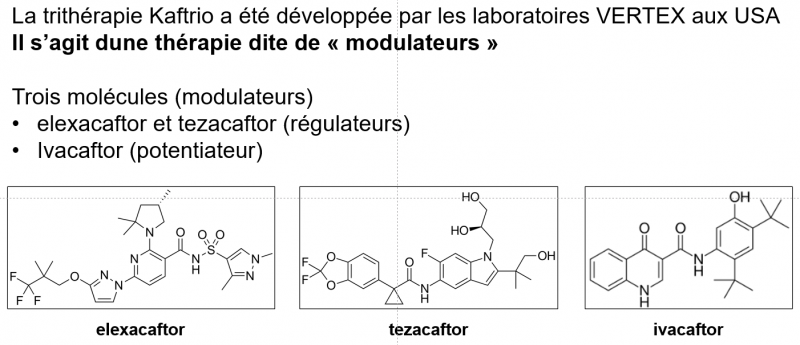
Et c’est ainsi que plusieurs molécules, dont elexacaftor, tezacaftor et ivacaftor ont démontré leur potentiel thérapeutique. L’elexacaftor et le tezacaftor (correcteurs), permettent d’augmenter fortement l’acheminement de la protéine mutée vers son site d’action sur les membranes cellulaires. L’ivacaftor (potentiateur) se lie sur la protéine mutée et lui rend son activité biologique (ouverture du canal anionique).
Ces trois molécules qualifiées de « modulateurs » de la protéine CFTR, et dont les effets se conjuguent,
sont associées dans le Kaftrio pour le traitement de la mucoviscidose
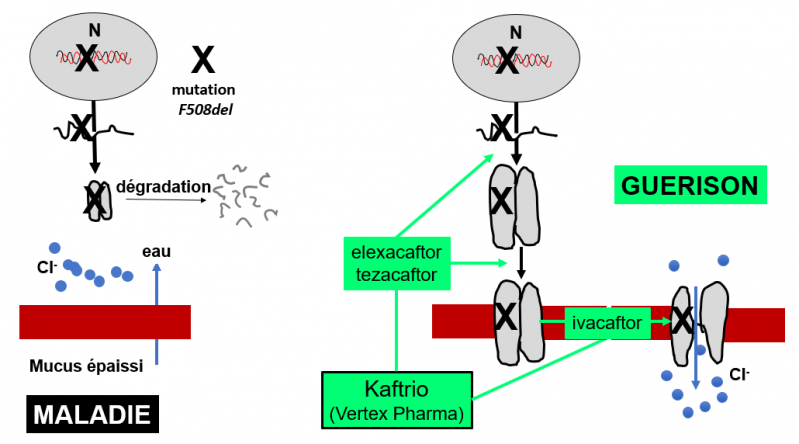
La trithérapie Kaftrio.
A gauche la situation chez les malades. A droite, la situation chez les malades traités par le Kaftrio. En se fixant sur la protéine CFTR, les deux "correcteurs" elexacaftor et tezacaftor inhibent la dégradation de la protéine et rétablissent son transport vers la membrane. De plus, après fixation sur la protéine CFTR, l'ivacaftor corrige sa conformation anormale (canal fermé) vers sa conformation fonctionnelle (canal ouvert). La situation normale est rétablie.
Cette trithérapie de "modulateurs" extrêmement prometteuse, développée par les Laboratoires Vertex aux Etats Unis, a été approuvée par la Food and Drug Administration américaine en 2019. En France depuis 2020, la Haute Autorité de Santé (HAS) autorise l’accès au Kaftrio pour les enfants homozygotes pour la mutation F508del et hétérozygotes pour la mutation F508del et porteurs d’une autre mutation du gène CFTR à fonction minimale (c’est à dire une mutation se manifestant par une perte de l’activité de la protéine CFTR). Cette autorisation de traitement d’abord accordée en 2020 uniquement aux enfants de 12 ans et plus, a été étendue à l’ensemble des enfants de 2 ans et plus en septembre 2023.
Certains variants ne sont malheureusement pas éligibles pour cette trithérapie, les protéines CFTR mutées qu’ils produisent y étant insensibles. C’est vers des traitements adaptés à ces variants que les recherches vont devoir se focaliser.
Cette trithérapie a donné un nouveau souffle de vie à 90 % des personnes atteintes de mucoviscidose porteuses d’au moins un variant F508del et devrait faire passer leur espérance de vie à plus de 80 ans comme vient de le montrer une publication récente (Lopez 2023).
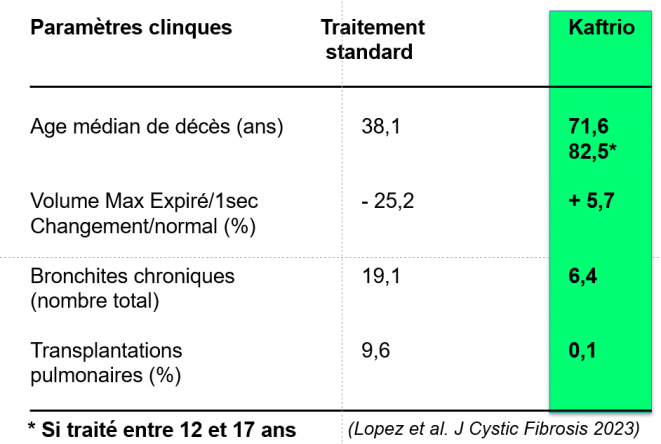
Ces chercheurs ont utilisé un modèle de micro simulation prédictif qui permet d'évaluer pour chaque malade, et dans le cadre d’essais cliniques contrôlés, les effets du Kaftrio à partir de données cliniques individuelles sur la survie, prenant en compte guérison, nombre de troubles pulmonaires sévères et nombre de transplantations pulmonaires, par rapport à d’autres traitements, notamment ceux administrés avant les années 2020. Les résultats résumés sur le tableau ci-dessus, montrent que pour les malades homozygotes pour la mutation F508del la survie est de 71,6 ans et elle passerait à 82,5 ans lorsque le traitement débute entre 12 et 17 ans. Cela représente une augmentation de plus de 33 ans d’espérance de vie par rapport à la thérapie classique utilisée avant les années 2020. Ce prolongement de la durée de vie des malades s'accompagne d'une amélioration très nette sur divers paramètres cliniques tels que le volume maximal d'air expiré par seconde, le nombre des bronchites sévères chroniques et le nombre des transplantations pulmonaires.
« Alors que les personnes atteintes de mucoviscidose étaient vraiment en difficulté il y a encore 5 ans, aujourd’hui elles reprennent le travail et réfléchissent à ce qu'elles pourraient faire pour leur retraite », confie le docteur Francis Collins, directeur de l’Institut National Américain de recherche sur le génome Humain.
Cependant, il faudra attendre encore quelques années avant de voir cette prédiction se réaliser car des incertitudes demeurent quant à l'impact de la maladie sur les possibles corrections apportées par le Kaftrio (réversibles versus irréversible ?) et notamment l'impact de l'âge du malade au moment de la mise en place du traitement (jeunes enfants versus adulte). A suivre donc...
Bibliographie
Ferrari 2017 : https://www.ncbi.nlm.nih.gov/pubmed/28079883
Esposito 2016 : https://www.ncbi.nlm.nih.gov/pubmed/26976279
Farrell 2018 : https://pubmed.ncbi.nlm.nih.gov/30089827/
Lopez 2023 : https://pubmed.ncbi.nlm.nih.gov/36849331/
Massie 2016 : https://www.ncbi.nlm.nih.gov/pubmed/27586437
Martiniano 2016 : https://www.ncbi.nlm.nih.gov/pubmed/27031658
Ong 2023 : https://pubmed.ncbi.nlm.nih.gov/37278811/
Quon 2016 : https://www.ncbi.nlm.nih.gov/pubmed/27030675
Maiuri 2015 : https://www.ncbi.nlm.nih.gov/pubmed/26046070
Wiuf 2001 : https://www.ncbi.nlm.nih.gov/pubmed/11556136
Date de dernière mise à jour : 13/01/2024
Ajouter un commentaire