
Syndrome de Rett
Cette maladie a été décrite pour la première fois en 1966 par un médecin Autrichien, Andreas Rett. Dans plus de 95% des cas, le gène impliqué est MECP2 (methyl-CpG-binding protein 2) localisé sur le chromosome X en position Xq28. Il comprend 10 505 paires de bases et est constitué de 4 exons. Ce gène code pour la protéine MeCP2 qui comprend 498 acides aminés. Cette protéine joue un rôle majeur dans la régulation épigénétique de nombreux gènes et dans d’autres fonctions. Les mutations responsables de la maladie apparaissent spontanément (mutations de novo) dans la lignée germinale paternelle (spermatozoïdes, voir ci-dessous, pères non porteurs) (Ehrhart 2016). Les filles sont quasi exclusivement touchées.
La maladie entre dans la catégorie des désordres neurologiques et apparaît très hétérogène dans ses manifestations. Elle altère le développement du système nerveux central. Elle se caractérise par une régression rapide des acquis après 6 à 18 mois de développement normal, avec une déficience intellectuelle sévère, des troubles respiratoires et cardiovasculaires, des crises d’épilepsie et/ou des convulsions, des scolioses, des troubles gastro-intestinaux, des troubles du sommeil et peut aussi revêtir diverses formes similaires à l’autisme. Les manifestations les plus fréquentes sont les mouvements stéréotypiques des mains, le langage absent ou rudimentaire, la marche instable ou jamais acquise. Une croissance ralentie du périmètre crânien correspondant à une atrophie cérébrale diffuse (notamment de la substance grise) peut être observée. Curieusement, ces symptomes majeurs de la maladie sont réversibles dans des modèles animaux (Guy 2007). La survie à l’âge de 25 ans est de 78%, et passe à 60% à 37 ans. Les causes de décès les plus fréquentes sont respiratoires (infections, asphyxie, détresse respiratoire).
Pourquoi seulement les filles ?
Nous possédons 22 paires de chromosomes dits “autosomes” et une paire de chromosomes dits “sexuels” (X et Y). Pour chaque paire, un chromosome provient du père et l’autre de la mère. La détermination du sexe réside dans la nature des chromosomes sexuels : la femme possède deux chromosomes X (l’un provenant de la mère et l’autre du père), et l’homme possède un X (provenant de la mère) et un Y (provenant du père). Le gène MECP2 qui se trouve impliqué dans la maladie de Rett se trouve sur le chromosome X. Mais, fait très important, la mutation n’est pas présente chez les parents qui ne sont pas malades (ni porteurs). La mutation dite “de novo” sur le gène MECP2 se produit spontanément et quasi exclusivement dans les spermatozoïdes (produits par le père). En effet, du fait de la multiplication cellulaire importante de la lignée paternelle (spermatogonies), la probabilité de présence de mutations sur l'ADN de ces cellules est élevée. Ainsi, le syndrome de Rett touche quasi exclusivement les filles, les garçons recevant leur chromosome X (porteur du gène MECP2 normal) de la mère (Trappe 2001; Girard 2001), comme le montre le schéma ci-dessous.
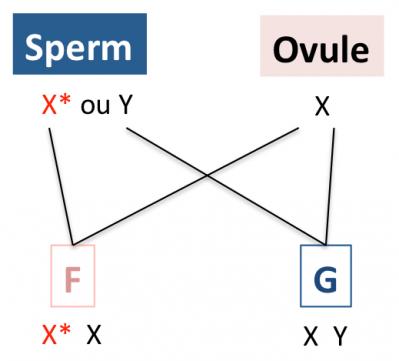
Lors de la formation de l’embryon, un ovule porteur du chromosome X est fécondé par un spermatozoïde porteur d’un chromosome X portant la mutation (X*) ou d’un chromosome Y. Dans la maladie de Rett, aucun des deux parents n’est malade. La mutation sur MECP2 porté par le chromosome X apparaît de façon spontanée dans les spermatozoïdes (X*). La fille F (malade) est issue de la fécondation d'un ovule (dont l'X est porteur du gène MECP2 normal) et d'un spermatozoïde porteur de MECP2 muté (X*). De fait, les garçons G (XY) ne sont que très rarement touchés, contrairement aux filles. La possibilité que l’ovule soit fécondé par un spermatozoïde X normal (sans mutation, pour donner une fille non malade) n’est pas représentée sur ce schéma.
Il est cependant possible, bien que cela soit très rare, que l'allèle MECP2 muté soit présent dans le chromosome X de l’ovule fécondé. Dans ces cas, les garçons issus de la combinaison X*/Y meurent d’une encéphalopathie précoce, très rapidement après la naissance, du fait de la totale absence de MeCP2 car ils ne possèdent qu’un seul chromosome X.
Régulation de MECP2
Chez la femme et comme pour tous les gènes portés par le chromosome X, une des deux copies de MECP2 est inactivée dans le chromosome X correspondant, afin d’éviter un phénomène de surdosage. L’expression du gène MECP2 et de la protéine MeCP2 est très étroitement régulée par divers facteurs transcriptionnels (SP1, SP3, C/EBP, CTCF, E2F1) et des microRNAs.
Les fonctions de MeCP2
Le gène MECP2 code pour deux protéines, MeCP2 e1 (exons 1, 3 et 4) et MeCP2 e2 (exons 2, 3 et 4). Ces deux formes sont identiques excepté dans leur partie C-terminale. La forme e1 semble plus spécifiquement impliquée dans la maladie car son niveau d’expression dans le cerveau est 12 fois superieur à celui de la forme e2 et son invalidation chez l’animal conduit au syndrome de Rett. Elle est également présente à un fort niveau dans le poumon et la rate, bien qu’on la retrouve (mais à des niveaux d’expression plus faibles) dans quasiment tous les tissus.
MeCP2 est une protéine multifonctionnelle, présente dans le noyau des cellules. Elle contrôle (en association avec le facteur de transcription CREB1) l’expression d’un grand nombre de gènes et le métabolisme à divers niveaux. La principale fonction de MeCP2 est épigénétique : la protéine se fixe spécifiquement sur les bases cytosines méthylées de l’ADN (5-Me-Cyt). Un pour cent de l’ADN humain est méthylé et les sites de méthylation se trouvent dans des régions à fort taux en CG, appelées îlots CpG. De tels îlots sont présents dans le promoteur de 60% de nos gènes. Il a été estimé que 20 millions de molécules MeCP2 sont présents dans le noyau des neurones (Skene 2010), un nombre équivalent à celui des marques 5-methyl-cytosine sur l’ADN, suggérant ainsi que chaque marque est couverte par une molécule de MeCP2.
La méthylation de l’ADN est une des composantes de la régulation épigénétique de l’expression des gènes. Les facteurs transcriptionnels qui contrôlent l’expression des gènes (accélération le plus souvent ou ralentissement parfois) ne peuvent se lier sur l’ADN méthylé de sorte que la méthylation est en général un inhibiteur de l’expression des gènes. De plus, l’ADN méthylé est la cible des histone-deacétylases (HDAC) dont l’action contribue à renforcer le compactage de l’ADN, reduisant encore l’accès aux facteurs de transcription.
Donc en bref, la protéine MeCP2 a pour fonction normale de moduler l’expression de nombreux gènes.
MeCP2 intervient ainsi dans diverses autres fonctions que l’on trouve altérées dans la maladie :
- le processus d’épissage des ARN messagers. Processus essentiel dont les perturbations du fait de mutations sur MECP2 peuvent conduire à la production d’ARN messagers anormaux et donc traduits en protéines anormales.
- La régulation de nombreux micro ARN dont l’expression est contrôlée au niveau épigénétique. Leur expression peut être augmentée ou diminuée en cas de mutation sur MECP2.
- La maturation des neurones, leur différenciation, leur morphologie, et le fonctionnement des synapses.
- La synthèse des protéines, qui se trouve réduite dans les cellules déficientes en MeCP2.
Les mutations dans le gène MECP2
Etant donné le rôle central de MeCP2 dans les diverses fonctions répertoriées ci-dessus, il est évident que toute mutation qui affectera l’activité ou l’expression de la protéine aura des répercussions importantes pour la vie du porteur. La sévérité de la maladie varie très largement d’une malade à l’autre, et les diverses mutations conduisent à des degrès divers de sévérité de la maladie (Cuddaph 2014). Il faut noter que toutes les mutations dans MECP2 ne causent pas la maladie, et que dans 5% des cas, la maladie peut être présente sans qu’il y ait de mutation sur MECP2. Certaines mutations de MECP2 causent des formes atténuées de la maladie, et des mutations dans d’autres gènes comme CDKL5 et FOG1 sont responsables de formes de maladie similaires au syndrome de Rett.
La majorité des mutations présentes chez les malades affectent l’interaction de la protéine MeCP2 avec l’ADN (MBD, figure ci-dessous) ou avec un complexe protéique represseur de l’expression de nombreux gènes, formé par le NCoR (Nuclear Corepressor Receptor) et le SMRT (Silencing mediator of Retinoic acid and thyroid receptor) (NID, voir la figure ci-dessous). Cette interaction avec ce complexe se fait spécifiquement par l’intermédiaire d’une région de MeCP2 qui comporte des acides aminés dont la mutation chez les malades, rend cette interaction impossible. A noter que des mutations dans le gène TBLR1 (un des composants du complexe NcoR/SMRT) qui sont associées à des déficiences intellectuelles rencontrées chez les malades, se traduisent également par une perte de l’interaction entre MeCP2 et le complexe NcoR/SMRT (Kruusvee 2017). Cette interaction semble donc cruciale pour un fonctionnement optimal du cerveau.

Cette figure montre en A le locus MECP2 sur le chromosome X (entre les gènes RCP et IRAK1). B : l’ARN prémessager MECP2e2 qui sera traduit en protéine MECP2e2 (exons 2, 3 et 4). La flèche sur l’exon 2 indique la position d’initiation de la traduction. C : l’ARN prémessager MECP2e1 qui sera traduit en protéine MeCP2e1 (exons 1, 3 et 4). La flèche sur l’exon 1 indique la position d’initiation de la traduction. La protéine MeCP2e1 est la protéine majoritairement exprimée dans le cerveau. D : schéma de la protéine MeCP2e1 avec les trois domaines fonctionnels, MBD (Domaine de liaison à l’ADN methylé), TRD (Domaine represseur de la transcription des gènes), et NID (Domaine de liaison au complexe NCoR/SMTR). Les mutations le plus souvent associées au syndrome de Rett sont indiquées au dessous de l’astérisque rouge qui donne leur position dans la protéine.
Les mutations p.Arg106Trp (remplacement de l’acide aminé arginine, Arg, en position 106 par l’acide aminé tryptophane, Trp), p.Arg168X (le X ici signifie que le codon pour l’acide aminé Arg de la forme sauvage en position 168 est remplacé par un codon stop, conduisant à une protéine tronquée de 167 acides aminés au lieu des 498 attendus) p.Arg255X, p.Arg270X, sont associées aux formes les plus sévères de la maladie. Les mutations p.Arg133Cys, p.Arg294X, p.Arg306Cys, et délétions en 3’, donnent des phénotypes moins sévères de la maladie ou des formes de maladies similaires au syndrome de Rett (Cuddaph 2014). Ces mutations se retrouvent chez environ deux tiers des malades (Leonard 2016).
Thérapie
Il n’y a pour l’heure pas de traitement définitif (étiologique) de cette maladie. Cependant, diverses stratégies thérapeutiques sont envisagées (en préclinique sur modèles animaux) ou déjà en cours (essais cliniques) dans le but de corriger les conséquences du déficit en protéine MeCP2 au niveau des circuits neuronaux, de certains facteurs de croissance neuronaux, de voies métaboliques, de synthèse des protéines et des mitochondries (Leonard 2016 ; Katz 2016):
i) préclinique : Memantine, Benserazide, Clenbuterol, Citalopram, LP-211, NLX101, L-838,417, Midazolam, NO-711, Acetyl-L-carnitine, Choline, Glatiramer acetate, Mecasermin, Fluvastatin
ii) en essai clinique : Dextromethorphan, Ketamine, Desipramine, Sarizotan, Fingolimod, Trofinetide, IGF1 humain recombinant, Lovastatine, Triheptanoin.
A côté de ces possibilités, d’autres stratégies plus radicales visant les causes de la maladie sont envisagées et actuellement testées en préclinique : thérapie génique, édition génomique, activation du gène MECP2 sur le chromosome X inactivé, utilisation de la protéine MeCP2 recombinante.
Protéine MeCP2 et autres maladies
Il apparaît que la protéine MeCP2 joue un rôle plus ou moins décisif dans d’autres maladies: Alzheimer, épilepsie, troubles de l’attention, maladie de Huntington, trisomie-21, etc. Les recherches effectuées pour le traitement du syndrome de Rett pourraient ainsi avoir des retombées importantes sur ces maladies qui représentent également un grand problème de santé publique. L’expression de la protéine MeCP2 est diminuée dans ces pathologies.
Bibliographie
Date de dernière mise à jour : 11/12/2019
Ajouter un commentaire